
Congenital disorders
A prospective cohort study protocol: monitoring and surveillance of adverse events following heterologous booster doses of Oxford AstraZeneca COVID-19 vaccine in previous recipients of two doses of Sinopharm or Sputnik V vaccines in Iran
Jul
Objectives
General objective
Measuring safety of the booster doses of AstraZeneca vaccine in people vaccinated with two doses of Sinopharm or Sputnik V vaccines in Iran.
Specific objectives
-
1)
To Estimate the incidence of different types of serious adverse events (SAEs) within 13 weeks in all enrolled vaccinated individuals after either the first or second booster vaccination with AstraZeneca.
-
2)
To estimate the incidence of different types of adverse events of special Interests (AESIs) within 13 weeks.
-
3)
To estimate the incidence of COVID-19 after either the first or second booster dose of A in all enrolled people.
Design
The present study is an active COVID-19 vaccine safety surveillance through an observational prospective cohort study that will be conducted in vaccination centers in Iran. The present protocol has been developed based on the COVID-19 vaccine safety guidance manual introduced by the WHO in 2020 [11].
Study settings
The study will be conducted in all major cities of Iran. Study sites are vaccination centers where the AstraZeneca is administered to the cohort population. Although vaccination against COVID-19 will be done in all health care centers plus other temporary locations, AstraZeneca has been provided in some predefined centers. In each site studied except Tehran, there will be a vaccination center to collect data on people get vaccinated by Astrazeneca. In Tehran, three vaccination centers have been determined to collect such data. Totally, we will collect data in all major cities. Study sites and vaccination centers will be selected by the Ministry of Health and Medical Education (MOHME). In order to receive the previous history of vaccination, we get access to Integrated Public Health System (IPHS). Table 1 shows characteristics of the cohort sites in the present study.
Study period
The time point for enrolment will be either the first or second booster vaccination with AstraZeneca in one of the study sites, after primary vaccination with Sinopharm or Sputnik V.
Each individual will be followed-up for 13 weeks after either the first or second booster vaccination with AstraZeneca vaccine. Thirteen weeks follow-up period was chosen because this covers the most common risk windows for AESIs (42 days).
Participants
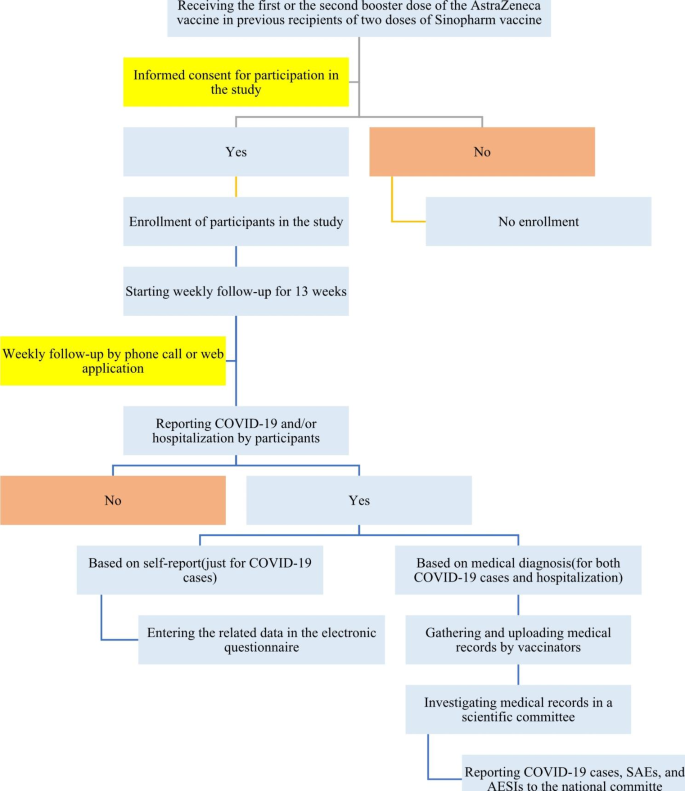
The study population includes all enrolled individuals who have received two doses of Sinopharm vaccine and either the first or second booster dose of AstraZeneca according to the national guidelines for immunization against COVID-19 in Iran in 2023. In addition, those who have been vaccinated with two doses of Sputnik V and received the first or second dose of AstraZeneca recruited. Study participation is strictly voluntary, and signed informed consent is obtained from all participants. The process of including participants in the present cohort study is shown in Fig. 1.
The flow chart of including participants in the present cohort study
Inclusion criteria
-
Written informed consent.
-
Willingness to continue to participate in the study until 3 months.
-
Enrolled individuals who have received the primary vaccination with two doses of Sinopharm or Sputnik V and either the first or second booster vaccination with AstraZeneca vaccine.
Exclusion criteria
-
Individuals unable to comply with study procedures (e.g. People with intellectual disabilities).
-
Individuals who have received booster vaccination by AstraZeneca neither as the first nor as the second booster vaccination.
-
Those who are less than 18 years old.
Sample size estimation
Regarding the WHO’s protocol, the sample size required to rule out an event with a frequency of 1 per 3,333 with a 95% confidence interval is 10,000 per vaccine [11]. Therefore, we are planning to include 20,000 and 10,000 eligible people with previous vaccination with Sinopharm and Sputnik V respectively. Regarding the higher proportion of immunization coverage for the Sinopharm vaccine compared to Sputnik V in Iran, we will include more vaccinated people with the Sinopharm vaccine in the present study. Participants will be considered to have completed the study when they have completed the last questionnaire 13 weeks after the booster dose. The end of the study is defined as the point at which the last subject enrolled has reached the 3 months follow-up period. In this study, convenience sampling will be used to include eligible participants until achieving the required sample size.
Subject recruitment and follow up
People who approach to one of the vaccination centers for booster vaccination, if they are eligible to be included, they will be consulted and get informed about the project. If they accept to participate, they will be asked to sign the consent form. On a day of vaccination, all the required information will be recorded on a web-based questionnaire. For weekly follow up, the study staff will call the participant by phone to collect data if the participant do not use the web application for self-reporting of adverse event. After two unsuccessful attempts to contact the participant by telephone, their next of kin will be contacted. After two unsuccessful attempts to contact their next of kin, the participant will be considered lost to follow-up if they do not complete the questionnaire by self-report. In fact, the participants are able to provide self-reported answers to all web-based questions using the mobile phone or a computer. The attempts to contact will be documented. We will record the underlying reason for withdrawal. Withdrawn participants and those lost to follow-up will not be replaced after the enrolment period has ended.
To identify SAEs and AESIs, each individual will be followed-up for 13 weeks after the booster dose of the AstraZeneca vaccine. The diagnoses reported by the participants during follow-up will be examined by a scientific committee including specialists in the field of infectious diseases, epidemiology, virology/immunology, internal diseases and others if needed. Such committees have been established by each university and they are involving in confirming the diagnosis for all SAEs and AESIs. In addition, there is a national committee to investigate adverse events related to vaccines, and therefore for all SAEs and AESIs, the patients’ files will be discussed in such committee. Such investigation helps participants to be treated within a standard protocol under the supervision of a scientific group.
As per WHO recommendation, all pregnant women inadvertently exposed to COVID-19 vaccine will follow up monthly until delivery, and the pregnancy outcome will be documented if it is possible. Although the available evidence does not fully support the use of AstraZeneca among pregnant women, we do not exclude such participants if they oblige to get a booster vaccination with AstraZeneca. The following maternal AESIs will be investigated among pregnant women: Maternal death and hospitalization, maternal thrombotic events, hypertensive disorders of pregnancy, miscarriage/spontaneous abortion, stillbirth, preterm birth, neonatal death, microcephaly, major congenital anomalies, infant death.
Study variables
Our independent variables include:
-
Age: Calculated as difference between enrolment date and the date of birth for people who will participate in the study.
-
Gender (Nominal variable): Male/Female.
-
Education (Ordinal variable): Total number of years of education.
-
Self-reported underlying health condition (Binary variable: yes/no): An underlying health condition is a chronic or long-term illness, which in turn weakens the immune system.
-
Self-reported weight (kg): The body weight for each respondent in our data set measured in kilogram.
-
Self-reported height (cm): The body weight for each respondent in our data set measured in centimeter.
-
Place of residence: The province where people live.
-
Exact date of pervious vaccinations.
Other main variables of the present study gathered in enrollment and follow-up questionnaires is shown in Table 2.
Exposure of interest
The exposure of interest is either the first or second booster vaccination with AstraZeneca after the primary vaccination with Sinopharm or Sputnik V. The COVID-19 vaccine brand, dose, vaccination date and batch number will be recorded.
Study outcomes
Serious adverse events (SAEs): hospitalization, death
Serious adverse events (SAEs) is defined as any over-night hospitalization or death. SAEs that lead to in-patient hospitalizations will be reported by the participant or their next of kin, and SAEs that results in death will be reported by their next of kin [12].
Adverse events of special interest (AESI) resulting in hospitalization
According to the WHO’s protocol template, the list of AESIs (Table 2) comprises events that have a proven relationship with immunization or a theoretical concern according to immunopathogenesis of COVID-19 disease [12].
Sever COVID-19
Severe COVID-19 disease is defined as COVID-19 disease resulting in blood oxygen saturation < 90%, signs of pneumonia and signs of severe respiratory distress. Occurrence of COVID-19 disease will be solicited throughout follow-up to collect information on laboratory-confirmed diagnosis or not [12]. In the present study, the occurrence of COVID-19 is based on clinical manifestations diagnosed by health professionals or/and the polymerase chain reaction (PCR) test.
Data management
We will develop a data management plan (DMP) before data collection begins and will describe all functions, processes, and specifications for data collection and cleaning. Study staff will enter data in an electronic questionnaire at several time points. The study staff will enter the participants’ information, contact details and data on the exposure at the time of vaccination and all other covariates.
Data security
In the present study, we stored the key-coded data in a secured database located in Iran. Data will be handled in line with all applicable data protection and privacy laws. No unauthorized persons will have access to the data. Data will be archived for 10 years, as per national regulations, and will then be destroyed. Data will be stored in a secure server environment and will be provided to researchers with an approved proposal by the project data manager. Analyses will be done on de-identified data by authorized researchers.
Source documents
The data sources for the exposure of interest will be participants and registered data. We should note that by adding the National code for each participant, all required information regarding the previous history of vaccination will be added to the web based questionnaire. The data source for covariates will be the participants as well. The data sources for the study outcomes will be the questionnaires completed by the participants (or their next of kin) as well as documentation from hospital and the universities committees as well as the national committee.
Data retention and archiving
Documents that individually and collectively permit evaluation of the study conduct and the quality of the data produced will be retained for 10 years in accordance with good pharmacoepidemiological practice guidelines. This will include the analytical data, analysis programs, and all output generated.
Quality assurance, monitoring, and reporting
For each province, a Co-PI will be selected who has direct communication with the PI and the central team. Remote and on-site monitoring of the study conduct will be performed throughout the study period to assess the accuracy and completeness of the data. The study site may be subject to a quality assurance visit. If so, the site will be contacted in advance to organize a monitoring visit. The investigator and site staff will assure direct access to all study documents for quality assurance monitors. The central team, included the authors of the present study, will control the quality of data once a week to identify errors, outliers and missing data. Furthermore, we will extract 1% of data randomly and contact participants to make sure they are adequately informed about the present study and know how to complete web based follow-up questionnaires. The examination of medical records will be conducted in the KUMS and the national committees. The committees will investigate medical records to approve or reject the association of the booster dose with a health outcome.
Interim analyses and reporting
For the study outcomes, interim analyses will be performed monthly but the formal report will be released within 1.5, 3, and 6 months. Such reports will be disseminated to all formal organizations and Iranian medical universities (including the Iranian Ministry of Health and Medical Education and WHO by request). In addition, the report will be published in relevant medical journals.
Final analyses and reporting
Final analyses will be conducted and a full study report will be written within 4 weeks after database lock. Study results will be shared with the national regulatory authorities for regulatory review, and with the national immunization program to inform public health policy decisions.
Study management
This study will be conducted by the principal investigator, Co-PIs. In addition, scientific advisors contribute to the development of materials, recruitment, training and management of sites, the electronic data capture, and data management and analyses. The Investigator and all study staff will conduct the present study in line with the ethics protocols of the Iran National Committee for Ethics in Biomedical Research. All human resources involved in the conduct of this study will be qualified by education, training, and experience to accomplish their tasks. For the purpose of this study, as well as in person communication with the PI and the central team, the co-PI and all other recruitment staff will take part in two separate training sessions held by the central team and a team from Iranian CDC.
Statistical analysis
In the present study, the data on SAEs and AESIs will be included for the whole cohort population. People who do not report any event(s) will be considered as participants without event(s). As AESIs can occur even after 42 days following the first booster dose, the sensitivity analysis will be done excluding those AESIs occurring within the first 12 days after the second booster dose.
-
We will calculate the frequency and proportion of participants reporting AESIs and SAEs by time since vaccination. For the proportions, 95% confidence intervals will be calculated using an exact method.
-
Observed-to-expected analyses will be conducted for SAEs and AESIs with an unknown risk window (most likely situation). The observed incidences for AESIs will be compared with background rates from the most appropriate sources. The expected rate will be age-stratified, and the standardized incidence ratio (SIR) will be calculated.
-
Incidence rate of severe COVID-19 disease (any COVID-19 disease diagnosed by a healthcare professional, laboratory-confirmed COVID-19, hospitalization for COVID-19, COVID-19 requiring intensive care unit (ICU) admission, COVID-19 disease resulting in death) will be calculated after either the first or the second booster dose of the AstraZeneca and within the period of follow-up.
The present study will be performed in line with the international ethical guidelines for epidemiology studies published by the Council for International Organizations of Medical Sciences (CIOMS), the Declaration of Helsinki and its amendments, good epidemiological practice (GEP) guidelines and the Iran National Committee for Ethics in Biomedical Research. Data protection and privacy regulations will be strictly observed in capturing, forwarding, processing, and storing individuals’ data. The study protocol and informed consent forms will be reviewed and approved by the Iran National Committee for Ethics in Biomedical Research.