
Cancer and neoplasms
microRNA sequencing for biomarker detection in the diagnosis, classification and prognosis of Diffuse Large B Cell Lymphoma
Jul
Abstract
Despite being considered a single disease, Diffuse Large B Cell Lymphoma (DLBCL) presents with variable backgrounds, which results in heterogeneous outcomes among patients, with 40% of them still having primary refractory disease or relapse. Thus, novel biomarkers are needed. In addition, multiple factors regarding its pathogenesis remain unclear. In this context, recent investigations point to the relevance of microRNAs (miRNAs) in cancer. However, regarding DLBCL, there is inconsistency in the data reported. Therefore, in this work, the main goals were to determine a miRNA set with utility as biomarkers for DLBCL diagnosis, classification, prognosis and treatment response, as well as to decipher the mechanism of action of deregulated miRNAs in the origin of the disease. We analyzed miRNA expression in a cohort of 78 DLBCL patients and 17 controls using small RNA sequencing and performed a miRNA-mRNA interaction network analysis. This way, we were able to define new miRNA expression signatures for diagnosis, classification, treatment response and prognosis, and we identified plausible mechanisms of action by which deregulated miRNAs could be involved in DLBCL pathogenesis. In summary, our study remarks that miRNAs could play an important role in DLBCL.
Introduction
Diffuse Large B Cell Lymphoma (DLBCL) is an aggressive type of non-Hodgkin lymphoma that represents the most common lymphoid malignancy in adults1. Despite the great heterogeneity observed among patients with DLBCL in terms of morphology, genetics, and biological behavior, most of them are treated with standard chemotherapy regimens, which allow complete remission in 75–80% of patients2,3. Nevertheless, the remaining patients will be refractory to first-line chemotherapy, and 30–40% will relapse after obtaining a complete remission. Therefore, the identification of new biomarkers for a better management of DLBCL patients is an urgent priority.
In this context, microRNAs (miRNAs), as post-transcriptional regulators of gene expression and biological functions, have shown potential as diagnostic, classification and prognostic predictors in cancer, although differences among studies were identified. For instance, miR-150-5p was downregulated in DLBCL in four studies4,5,6,7, while showed upregulated expression in another study8 and unchanged in another one9. Similarly, miR-222-3p was associated with good prognosis in four studies10,11,12,13 while no association was observed in other four studies6,9,14,15. For that reason, in a recently published systematic review16 we sought to identify a signature of miRNAs of relevance in DLBCL. However, we observed that the studies performed usually considered a limited set of selected miRNAs, which limits the number of comparable results and centers the discussion on those miRNAs that are better known, leaving other miRNAs aside. Therefore, we considered that it was necessary to perform large-scale studies with a wider array of miRNAs using techniques such as next-generation sequencing that allow a deeper and unbiased identification of miRNAs with the potential to be used as biomarkers in DLBCL. Moreover, this technology allows the characterization of the whole miRNAome, providing an insight in the global mechanism of action of deregulated miRNAs.
Consequently, the main goals of the present study were to determine a miRNA set with utility in DLBCL diagnosis, classification, prognosis and treatment response, through the analysis of all the expressed miRNAs in DLBCL using small RNA sequencing, as well as to decipher the mechanism of action of deregulated miRNAs in the origin of the disease through the development of a miRNA-mRNA interaction network.
Results
MiRNAs deregulated in DLBCL
A total of 1584 miRNAs were identified through the miRNA sequencing analysis. A principal component analysis (PCA) was performed including the expression of all the miRNAs to obtain an overview of the grouping of samples according to their variance. Patient and control samples cluster as clearly separate groups (Supplementary Fig. S1). To obtain a comprehensive list of miRNAs deregulated in DLBCL, we compared the expression of each miRNA in 78 DLBCL samples at diagnosis with 17 non-tumoral ganglia from individuals without DLBCL. We noted that 146 miRNAs exhibited statistically significant differences in expression between samples of DLBCL patients at diagnosis and control samples. Of these miRNAs, 122 exhibited increased abundance in DLBCL, while 24 miRNAs exhibited decreased abundance. The 20 most discriminatory up- and down-regulated miRNAs are listed in Table 1.
MiRNAs differentially expressed among DLBCL subtypes
By analyzing expression profiling of 32 Germinal center B-cell like (GCB) and 21 Non-GCB DLBCL samples at diagnosis, eight miRNAs were found to be differentially expressed. Five of them were upregulated in GCB DLBCL patients (miR-129-2-3p, miR-4464, miR-3150b-3p, miR-138-5p and miR-129-5p) and three miRNAs were upregulated in Non-GCB subtype (miR-511-5p, miR-205-5p, miR-3652) (Table 2).
MiRNAs associated with DLBCL treatment response
Eleven miRNAs were differentially expressed between samples at diagnosis from patients with long-term complete remission (n = 50) and refractory patients (n = 12), of which, 10 miRNAs were upregulated in patients with complete remission (miR-12136, miR-129-5p, miR-129-1-3p, miR-3150b-3p, miR-127-3p, miR-3681-5p, miR-370-3p, miR-4464, miR-129-5p and miR-3928-3p) and one miRNA was downregulated in the same group of patients (miR-192-5p) (Table 3).
MiRNAs as prognostic biomarkers in DLBCL and their impact on survival
MiRNA expression at diagnosis of 50 patients with long-term remission and 16 patients that relapsed within 10 years after diagnosis was compared. The results revealed that seven miRNAs (miR-4444, miR-449c-5p, miR-3681-5p, miR-3928-3p, miR-449b-5p, miR-370-3p, and miR-4424) were significantly upregulated in patients with long-term remission and three miRNAs (miR-133a-3p, miR-208b-3p, and miR-205-5p) were significantly upregulated in patients that relapsed (Table 4).
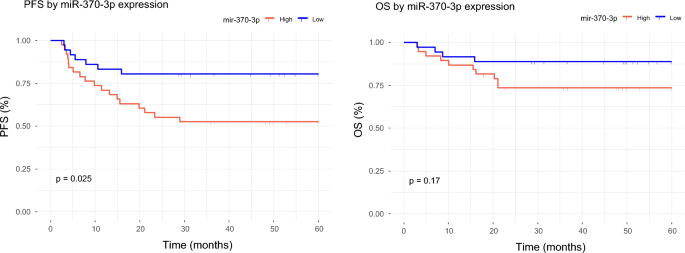
Patient survival curves were generated to represent 5-year progression-free survival (PFS) and overall survival (OS), dividing the patients into high-expression and low-expression groups according to the mean expression of each of the ten miRNAs significantly associated with prognosis. The long-rank test revealed that, among the miRNAs associated with good prognosis, high expression of miR-370-3p was associated with better 5-year PFS (OS p-value = 0.17, and PFS p-value = 0.025) (Fig. 1). We also performed Cox PH multivariate analysis including two established indicators of DLBCL patient outcome (COO-Subtype and IPI) as covariates. However, the results of this analysis revealed that miR-370-3p was not associated with PFS independently of IPI and subtype.
Survival analysis of miR-370-3p. The red line represents patients with high miR-370-3p expression and the blue line represents patients with low miR-370-3p expression.
miRNA-mRNA interaction network analysis
For the 122 upregulated miRNAs found in our study (DLBCL vs controls), we identified a total of 482 experimentally validated targets using MirTarbase. Regarding the 24 downregulated miRNAs from our study, a total of 375 experimentally validated target genes were identified. Analysis of the GSE56315 dataset identified through the search in the GEO, revealed a total of 1918 significantly upregulated genes and 4545 downregulated genes in DLBCL patients compared with healthy control individuals.
Integration of both datasets showed that downregulated miRNAs targeted a total of 138 genes among those highly expressed in DLBCL (Supplementary Table S1), and upregulated miRNAs a total of 76 targets among those genes downregulated in DLBCL (Supplementary Table S2). Some miRNAs showed interactions with several targets (Supplementary Table S3). For instance, miR-9-5p was shown to target 25 genes downregulated in DLBCL, and miR-146a-5p and miR-182-5p were shown to target 24 and 20 genes downregulated in DLBCL, respectively. Some target genes showed interactions with several miRNAs (Supplementary Table S4). For instance, FOXO1, BCL2, GS3KB and PTEN, downregulated in DLBCL, are targeted by 7, 5, 5 and 4 miRNAs deregulated in DLBCL, respectively. In order to evaluate the pathways that could be affected by the deregulated miRNAs through their target genes, we performed a pathway enrichment analysis using the ConsensusPathDB web tool. Among the most significantly over-represented pathways, we identified some involved in cancer, such as FoxO signaling pathway, signaling by Receptor Tyrosine Kinases, and PI3K-Akt signaling pathway (Fig. 2).

Genes of pathways in cancer targeted by downregulated and upregulated microRNAs (adapted from KEGG database).
Discussion
In this study, we identified new signatures of miRNAs of relevance in DLBCL with potential to improve diagnosis, subtype characterization and treatment response through small RNA sequencing. To our knowledge, few reports exist in which miRNA sequencing were used to identify miRNA signatures in cancer, and only one which analyzed miRNAs with Next Generation Sequencing (NGS) in DLBCL9. Notably, deregulation of most of these miRNAs had not been reported previously in DLBCL.
Regarding miRNAs deregulated at diagnosis in DLBCL, 146 miRNAs differentially expressed between DLBCL samples and controls were identified, pointing to a relevant role of miRNAs in the pathogenesis of the disease. Of note, all miRNAs we previously proposed as a consistent signature of deregulation in DLBCL in a systematic review (miR-150-5p, miR-146a-5p, miR-155-5p and miR-21-5p)16 were identified in the current study following a similar expression pattern. Moreover, we also identified other miRNAs highly deregulated in DLBCL. Indeed, regarding the most upregulated and downregulated miRNAs, only miR-210-3p, miR-146a-5p, miR-129-5p, miR-215, miR-150-5p, miR-224-5p, miR-335-5p, miR-145-5p, miR-497-5p, and miR-151b had been studied previously in DLBCL.
Comparing miRNA expression in DLBCL GCB and Non-GCB subgroups, eight miRNAs were identified with subtype differentiation potential. Five of them were upregulated in GCB DLBCL patients (miR-129–2-3p, miR-4464, miR-3150b-3p, miR-138-5p and miR-129-5p) and three were upregulated in Non-GCB subtype (miR-511-5p, miR-205-5p, and miR-3652). Of note, miR-28-5p, which was proposed as a biomarker of GCB DLBCL in our previous systematic review, pending further confirmation16, was also differentially expressed among subtypes, although the logFC did not reach the threshold set for the present study. Interestingly, higher levels of miR-129-5p and miR-138-5p in the GCB subtype had been previously reported6,9,14,17. As part of the miRNA-mRNA interaction network analysis, we observed that they could target transcripts that are known to be deregulated in the formation of germinal center lymphomas18, including genes related with the cell cycle (CDKN1A), MAPK and NFkB signaling (MAPK1). Interestingly, we also found TP53 as a target gene of miR-129-3p, miR-129-5p, and miR-138-5p. TP53 gene is a crucial tumor suppressor that mediates cell-cycle arrest, DNA repair, apoptosis, senescence, and autophagy19,20, and its dysfunction is implicated in lymphomagenesis and disease progression. Recent studies have shown that TP53 mutations have prognostic value in GCB-DLBCL, but not in Non-GCB-DLBCL21, which could suggest that GCB-DLBCL is TP53-dependent while Non-GCB DLBCL is TP53-independent, and thus, could support the role of miR-129-3p, miR-129-5p, and miR-138-5p in GBC subtype.
We also identified a predictive miRNA signature for therapy response, which included the upregulation of ten miRNAs (miR-12136, miR-129a-5p, miR-129-1-3p, miR-3150b-3p, miR-127-3p, miR-3681-5p, miR-370-3p, miR-4464, miR-129b-5p and miR-3928-3p) in association with good response and upregulation of miR-192-5p associated with chemoresistance to R-CHOP DLBCL treatment. Notably, deregulation profiling of most of these miRNAs in DLBCL had not been previously reported, with a limited number of studies carried out; but some of them, such as miR-192 had been studied in the context of other types of cancer22,23, supporting their role in drug response. Interestingly, miR-370-3p has been recently shown as a target of circ_0000877 and LINC00857 mediating their effect on DLBCL cells survival, which would be in agreement with its association with good response to therapy24,25. In addition to being putative biomarkers for R-CHOP treatment response prediction, these miRNAs could also be considered in the future as novel therapeutic targets for personalized miRNA-based therapy, which could be an alternative adjuvant therapy, although further studies are needed in this area of research before such therapies can be translated to the clinic.
In addition to treatment response, the ability to predict survival accurately may be crucial for initial treatment planning in patients with DLBCL. We identified seven miRNAs (miR-4444, miR-449c-5p, miR-3681-5p, miR-3928-3p, miR-449b-5p, miR-370-3p, miR-4424) significantly upregulated in patients with long-term remission and four miRNAs (miR-133a-3p, miR-133a-3p, miR-208b-3p, miR-205-5p) upregulated in relapsed patients. In addition, the survival analysis reveals that high expression of miR-370-3p is significantly associated with PFS. However, the association was not maintained when IPI and subtype were included in the model. As mentioned above, its role as a mediator of DLBCL cell survival in vitro has been stablished in two different studies. Thus, further studies are warranted to confirm its role as outcome predictor.
To gain insight into the global mechanism of action of deregulated miRNAs, we also performed a miRNA-mRNA interaction network analysis. Integration analysis determined that up to 214 genes could have altered expression in DLBCL because of miRNA deregulation. In this line, there were 138 genes with low expression in DLBCL that could be due, at least in part, to the increased expression of miRNAs identified in the current study. Moreover, 76 genes were highly expressed that could be due to a lack of regulation by the miRNAs lowly expressed here identified. Among the most interesting downregulated genes targeted by more than one miRNA overexpressed in DLBCL, we identified FOXO1 and PTEN, involved in PI3K-Akt pathway26, predicted to be targeted by 5 (miR-182-5p, miR-183-5p, miR-135a-5p, miR-9-5p, and miR-9-3p) and 4 (miR-155, miR-320a, miR-205 and miR-182-5p) overexpressed miRNAs, respectively. FOXO1 is a tumor suppressor and its downregulation leads to promoting tumorigenesis by favoring resistance to stress, proliferation, and increased cellular survival27. Abnormal cells that would normally undergo apoptosis may instead survive in the absence of FOXO1, which results in tumor expansion28. In fact, downregulation of FOXO1 has been previously reported in DLBCLs29. In the same line, PTEN is a tumor suppressor that acts upstream of FOXO1 negatively regulating AKT. Without PTEN regulation, activated AKT phosphorylates FOXO1, which is subsequently exported from the nucleus into the cytoplasm and degraded by the proteasome30. Interestingly, PTEN is the major negative regulator of the PI3K/AKT signaling pathway, which is constitutively activated in 25–50% of DLBCL according to recent studies31. Interestingly, miR-182-5p targeted both genes and could be a crucial miRNA modulating the PI3K/AKT pathway by targeting PTEN, upstream, and FOXO1, downstream. Further supporting our findings, a relation between miR-182 and miR-183 and FOXO1 suppression have been previously described in classical Hodgkin lymphoma32.
Regarding the upregulated genes in DLBCL that could be a consequence of miRNA downregulation, the most significant result is BCL2. Evidence revealed that elevated expression of anti-apoptotic members such as Bcl-2 is one of the major contributing factors to B cell lymphomagenesis33. Since then, many studies have determined that BCL2 is one of the most important oncogenes in cancer and that it is linked with lymphoma development, particularly when c-MYC is overexpressed34. This gene could be regulated negatively by miR-135a-5p, miR-234-5p, miR-139-5p, miR-451a, and miR-497-5p. Therefore, the low expression of the mentioned miRNAs observed in DLBCL would contribute to BCL2 overexpression. In fact, previous studies have confirmed that deletion or downregulation of miRNAs is a mechanism for BCL2 overexpression35.
Finally, this study has some limitations that need to be addressed. First, we analyzed miRNA expression from formalin-fixed paraffin-embedded (FFPE) blocks, in which RNA quality is usually compromised. However, compared to mRNA, miRNAs are relatively resistant to RNase degradation36, and previous studies demonstrated that miRNA expression from FFPE samples is in good correlation with fresh frozen samples37. Secondly, patients were diagnosed in three different hospitals with different follow up routines, which could be a source of heterogeneity, although the treatment protocols were similar. Finally, we also have to mention that in this study the Han’s IHC algorithm was used to classify the patients into GCB and Non-GCB subtypes. Even though studies have shown clearly that the IHC-based classification provided very similar outcome prediction compared with the gene expression profiling based classification, which is the gold standard38, variable reproducibility of IHC stains and interpretations, may be the reason for inconsistent results39.
In conclusion, in the present study, a comprehensive analysis of miRNA profiles by next-generation sequencing technology allowed us to identify signatures that could be of relevance in DLBCL. Prospective studies are warranted to further validate our findings and explore new therapeutic options based on the miRNA profile of each patient. Moreover, we explored the mechanisms by which deregulated miRNAs contributes to DLBCL pathogenesis, with miR-182-5p playing a key role in PI3K/AKT pathway.
Materials and methods
Population of study
Samples from 78 DLBCL patients were obtained at diagnosis from formalin-fixed paraffin-embedded (FFPE) tumor tissue (50 patients with long-term complete remission, 12 patients with refractory disease, and 16 patients that relapsed within 10 years after diagnosis). Samples were collected from 1999 to 2018 at the Hematology Units of 3 Spanish reference hospitals (Cruces University Hospital, Donostia University Hospital, and Araba University Hospital). Control samples were obtained from non-tumoral ganglia from individuals without DLBCL, which were collected at Araba University Hospital. Demographic and clinical data were obtained from patient’s medical files by two independent clinical researchers (Table 5). Demographic information was also obtained for the controls. All patients were treated with R-CHOP or similar chemotherapy regimens, which included rituximab. The study was approved by the Clinical Research Ethical Committee of the Basque Country (P2016121). Signed informed consent was obtained from each participant and the study was carried out according to the Declaration of Helsinki.
Sample processing, Small RNA-seq library preparation and sequencing
RNA was isolated from FFPE samples with the miRNeasy FFPE Kit (Qiagen, Hilden, Germany). One µg of total RNA was used for library preparation with TruSeq small RNA Sample Prep Kit (Illumina), according to the manufacturer’s protocol. Libraries were analyzed using Agilent DNA High Sensitivity chip and sequenced Single Read, 50nts (v4) on Illumina’s HiSeq 2500 with a depth of approximately 10 million reads per sample. All the process was carried out at the Centre for Genomic Regulation (CRG). The data are openly available in GEO database, reference number GSE18579640.
Bioinformatic analysis and differential miRNA expression
Reads were trimmed for the presence of the small RNA adapter using Skewer and filtered removing every sequence shorter than 15 and longer than 40 bases. The remaining ones were then aligned to the reference genome (GRCh38) using ShortStack and the resulting alignments used by htseq-count for determining the number of tags per gene. We used the annotation of the miRBase Sequence Database v22.1mirbase in order to select only miRNA genes. Every small RNA with less than 10 reads considering the sum of the reads in every condition was excluded, and we used the remaining genes for differential expression analysis with DESeq2. p-values were adjusted using False discovery rate (p-adj). Differentially expressed miRNAs were those with p-adj < 0.05 and a log2 fold change (logFC) > 2 or < − 2. Principal component analysis was performed using the prcomp package from R to analyze the transformed read counts of all miRNAs.
Survival analysis
Survival analyses were conducted to confirm the absence of effect of changes in chemotherapy (standard vs. intensified) on survival. Additional survival analyses were performed to estimate the effect on the progression-free survival (PFS) and overall survival (OS) of those miRNAs differentially expressed between samples at diagnosis from patients with long-term remission and those that relapsed. The median value of the expression of those differentially expressed miRNAs was considered to categorize patients into low or high expression groups. Kaplan–Meier analysis was used to define the survival curves and the log-rank test was used to assess significance. For the multivariate analysis, we also considered the subtype and IPI status using the Cox proportional hazards (Cox PH) method. All calculations were performed using the Survival R package. Statistically significant associations with survival were those with p-value < 0.05.
miRNA-mRNA interaction network
Using our own data on miRNA expression (DLBCL patients vs controls) and mRNA expression data contained in the Gene Expression Omnibus (GEO) database, we constructed a miRNA-mRNA interaction network to elucidate the mechanism of DLBCL development. First, for those miRNAs deregulated in patients compared to controls, target genes were identified using mirTarbase41 (http://mirtarbase.cuhk.edu.cn/php/index.php) to search for experimentally validated miRNA-mRNA interactions. For the current study, only strong miRNA-mRNA interactions were selected. Secondly, after a comprehensive search of databases in GEO (http://www.ncbi.nlm.nih.gov/geo/), the only microarray dataset (GSE56315) containing gene expression information of both DLBCL samples (n = 55) and healthy controls (n = 33) was downloaded42,43. Differentially expressed genes (DEG) were identified using GEO2R tool with default parameters. We established the following inclusion criteria for the DEGs: upregulated genes must have a logFC ≥ 2 and p-adj < 0.05, while downregulated genes must have a logFC ≤ − 2 and p-adj < 0.05. Finally, experimentally validated targets of differentially deregulated miRNAs were overlapped with mRNAs differentially expressed obtained from GEO databases as represented in Supplementary Fig. S2.
The intersection of the upregulated and downregulated genes was mapped using the Venn package (http://bioinformatics.psb.ugent.be/webtools/Venn/). Signaling pathway enrichment analysis (Consensus Pathway database http://cpdb.molgen.mpg.de/) were conducted with overlapping genes.
Data availability
The data are openly available in GEO database, reference number GSE185796.
References
-
Swerdlow, S. H. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–2390 (2016).
Google Scholar
-
Zhang, J. et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 110, 1398–1403 (2013).
Google Scholar
-
Coiffier, B. et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: A study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood 116, 2040–2045 (2010).
Google Scholar
-
Caramuta, S. et al. Role of microRNAs and microRNA machinery in the pathogenesis of diffuse large B-cell lymphoma. Blood Cancer J. 3, e152–e152 (2013).
Google Scholar
-
Fassina, A. et al. The miR-17-92 microRNA cluster: A novel diagnostic tool in large B-cell malignancies. Lab. Invest. 92, 1574–1582 (2012).
Google Scholar
-
Roehle, A. et al. MicroRNA signatures characterize diffuse large B-cell lymphomas and follicular lymphomas. Br. J. Haematol. 142, 732–744 (2008).
Google Scholar
-
Jia, Y. J. et al. HDAC6 regulates microRNA-27b that suppresses proliferation, promotes apoptosis and target MET in diffuse large B-cell lymphoma. Leukemia 32, 703–711 (2018).
Google Scholar
-
Handal, B. et al. Investigating the expression of oncogenic and tumor suppressive MicroRNA in DLBCL. J. Assoc. Genet. Technol. 39, 14–20 (2013).
Google Scholar
-
Lim, E. L. et al. Comprehensive miRNA sequence analysis reveals survival differences in diffuse large B-cell lymphoma patients. Genome Biol. 16, 18–18 (2015).
Google Scholar
-
Montes-Moreno, S. et al. miRNA expression in diffuse large B-cell lymphoma treated with chemoimmunotherapy. Blood 118, 1034–1040 (2011).
Google Scholar
-
Alencar, A. J. et al. MicroRNAs are independent predictors of outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Clin. Cancer Res. 17, 4125 (2011).
Google Scholar
-
Malumbres, R. et al. Differentiation stage–specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas. Blood 113, 3754–3764 (2009).
Google Scholar
-
Sun, S., Wang, H. & Ji, M. Overexpression of miR-222-3p promotes the proliferation and inhibits the apoptosis of diffuse large B-cell lymphoma cells via suppressing PPP2R2A. Technol. Cancer Res. Treat. 18, 1533033819892256 (2019).
Google Scholar
-
Lawrie, C. H. et al. Expression of microRNAs in Diffuse Large B Cell Lymphoma is associated with immunophenotype, survival and transformation from follicular lymphoma. J. Cell Mol. Med. 13, 1248–1260 (2009).
Google Scholar
-
Shepshelovich, D. et al. MicroRNA signature is indicative of long term prognosis in diffuse large B-cell lymphoma. Leuk. Res. 39, 632–637 (2015).
Google Scholar
-
Larrabeiti-Etxebarria, A., Lopez-Santillan, M., Santos-Zorrozua, B., Lopez-Lopez, E. & Garcia-Orad, A. Systematic review of the potential of MicroRNAs in Diffuse Large B Cell Lymphoma. Cancers (Basel) 11, 144 (2019).
Google Scholar
-
Iqbal, J. et al. Global microRNA expression profiling uncovers molecular markers for classification and prognosis in aggressive B-cell lymphoma. Blood 125, 1137–1145 (2015).
Google Scholar
-
Shaffer, A. L. 3rd., Young, R. M. & Staudt, L. M. Pathogenesis of human B cell lymphomas. Annu. Rev. Immunol. 30, 565–610 (2012).
Google Scholar
-
Levine, A. J. & Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 9, 749–758 (2009).
Google Scholar
-
Vousden, K. H. & Prives, C. Blinded by the light: The growing complexity of p53. Cell 137, 413–431 (2009).
Google Scholar
-
Young, K. H. et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: An international collaborative study. Blood 112, 3088–3098 (2008).
Google Scholar
-
Stasiłojć, T. G., Supernat, A., Jassem, J., Żaczek, A. J. & Bigda, J. MiR-192 and miR-662 enhance chemoresistance and invasiveness of squamous cell lung carcinoma. Lung Cancer 118, 111–118. https://doi.org/10.1016/j.lungcan.2018.02.002 (2018).
Google Scholar
-
Poel, D. et al. Expression of let-7i and miR-192 is associated with resistance to cisplatin-based chemoradiotherapy in patients with larynx and hypopharynx cancer. Oral Oncol. 109, 104851. https://doi.org/10.1016/j.oraloncology.2020.104851 (2020).
Google Scholar
-
Chengzhi, Z. et al. Hsa_circ_0000877 facilitates the progression of diffuse large B-cell lymphoma by miR-370-3p/mitogen-activated protein kinase kinase kinase kinase 4/Hippo pathway. Anticancer Drugs https://doi.org/10.1097/CAD.0000000000001366 (2022).
Google Scholar
-
Huang, Y., Lin, Y., Song, X. & Wu, D. LINC00857 contributes to proliferation and lymphomagenesis by regulating miR-370-3p/CBX3 axis in diffuse large B-cell lymphoma. Carcinogenesis 42(5), 733–741. https://doi.org/10.1093/carcin/bgab013 (2021).
Google Scholar
-
Ho, K. K., Myatt, S. S. & Lam, E.W.-F. Many forks in the path: Cycling with FoxO. Oncogene 27, 2300–2311 (2008).
Google Scholar
-
Myatt, S. S. & Lam, E. W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 7(11), 847–859. https://doi.org/10.1038/nrc2223 (2007).
Google Scholar
-
Reagan-Shaw, S. & Ahmad, N. The role of Forkhead-box Class O (FoxO) transcription factors in cancer: A target for the management of cancer. Toxicol. Appl. Pharmacol. 224, 360–368 (2007).
Google Scholar
-
Trinh, D. L. et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood 121, 3666–3674 (2013).
Google Scholar
-
Boccitto, M. & Kalb, R. G. Regulation of Foxo-dependent transcription by post-translational modifications. Curr. Drug Targets 12, 1303–1310 (2011).
Google Scholar
-
Uddin, S. et al. Role of phosphatidylinositol 3′-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood 108, 4178–4186 (2006).
Google Scholar
-
Xie, L. et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood 119, 3503–3511 (2012).
Google Scholar
-
Adams, J. M. & Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27–36 (2018).
Google Scholar
-
Strasser, A., Harris, A. W., Bath, M. L. & Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348, 331–333 (1990).
Google Scholar
-
Calin, G. A. et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. U. S. A. 105, 5166–5171 (2008).
Google Scholar
-
Li, J. et al. Comparison of miRNA expression patterns using total RNA extracted from matched samples of formalin-fixed paraffin-embedded (FFPE) cells and snap frozen cells. BMC Biotechnol. 7, 36–36 (2007).
Google Scholar
-
Xi, Y. et al. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA 13, 1668–1674 (2007).
Google Scholar
-
Choi, W. W. L. et al. A New immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin. Cancer Res. 15, 5494 (2009).
Google Scholar
-
Barton, S., Hawkes, E. A., Wotherspoon, A. & Cunningham, D. Are we ready to stratify treatment for diffuse large B-cell lymphoma using molecular hallmarks?. Oncologist 17, 1562–1573 (2012).
Google Scholar
-
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE185796 (2022).
-
Huang, H.-Y. et al. miRTarBase 2020: Updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 48, D148–D154 (2020).
Google Scholar
-
Dybkær, K. et al. Diffuse large B-cell lymphoma classification system that associates normal B-cell subset phenotypes with prognosis. J. Clin. Oncol. 33, 1379–1388 (2015).
Google Scholar
-
Nørgaard, C. H. et al. Subtype assignment of CLL based on B-cell subset associated gene signatures from normal bone marrow—a proof of concept study. PLoS ONE 13, e0193249–e0193249 (2018).
Google Scholar
Acknowledgements
This research was funded by the Basque Government (IT989-16 and IT1559-22) and EiTB maratoia (Bioef) (BIO15/CA/022/BC). AGC was supported by a postdoctoral fellowship from the Canadian Institute of Health Research (CIHR) (201910MFE-430442-73764).
Author information
Authors and Affiliations
Contributions
A.L.E., E.L.L. and A.G.C. conceived and designed the study; A.L.E., L.C., N.B.A. and A.G.C. performed the bioinformatic analysis; A.L.E., J.A.M. and E.L.L. performed genomic data analysis; M.M.A., A.G., I.R.D. and I.G. provided the samples and clinical data; I.M.G. and E.L.L. obtained the funding to perform this project; A.L.E., I.M.G., E.L.L. and A.G.C. wrote the first draft of the manuscript. All authors reviewed the manuscript and approved of the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Larrabeiti-Etxebarria, A., Bilbao-Aldaiturriaga, N., Arzuaga-Mendez, J. et al. microRNA sequencing for biomarker detection in the diagnosis, classification and prognosis of Diffuse Large B Cell Lymphoma.
Sci Rep 13, 12159 (2023). https://doi.org/10.1038/s41598-023-39271-7
-
Received: 18 October 2022
-
Accepted: 22 July 2023
-
Published: 27 July 2023
-
DOI: https://doi.org/10.1038/s41598-023-39271-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.