
Congenital disorders
Novel epigenetic molecular therapies for imprinting disorders
Aug
Abstract
Genomic imprinting disorders are caused by the disruption of genomic imprinting processes leading to a deficit or increase of an active allele. Their unique molecular mechanisms underlying imprinted genes offer an opportunity to investigate epigenetic-based therapy for reactivation of an inactive allele or reduction of an active allele. Current treatments are based on managing symptoms, not targeting the molecular mechanisms underlying imprinting disorders. Here, we highlight molecular approaches of therapeutic candidates in preclinical and clinical studies for individual imprinting disorders. These include the significant progress of discovery and testing of small molecules, antisense oligonucleotides, and CRISPR mediated genome editing approaches as new therapeutic strategies. We discuss the significant challenges of translating these promising therapies from the preclinical stage to the clinic, especially for genome editing based approaches.
Introduction
Genomic imprinting is a special form of epigenetic regulation, resulting in monoallelic gene expression depending on parent-of-origin. Genomic imprinting was first described in the early 1980s in mice from the elegant pronuclear transfer experiments by Surani and Solter [1, 2]. They showed that the contribution of the parental genomes to offspring genomic transcriptions were nonequivalent. Insulin-like growth factor 2 receptor (Igf2r) was the first imprinted gene discovered in mice in 1991 [3]. In humans, ~130 genes have been reported as imprinted genes and additional ~120 genes are predicted or provisioned (geneimprint.com). A number of imprinted genes are essential for normal embryonic development and neurodevelopment [4,5,6]. Several distinct features are associated with imprinted gene regulation [7,8,9,10]. First, the imprinted genes are frequently clustered and under a coordinated epigenetic regulation. Two major clusters of imprinted genes are in the chromosome 11p15.5 and 15q11-q13 regions. Second, the imprinted genes are frequently associated with allelic specific epigenetic modifications of DNA methylation, post-translational histone modifications, and chromatin structure [11,12,13,14]. Third, a significant number of imprinted genes are noncoding RNAs [15, 16]. Fourth, the antisense and long noncoding RNAs (lncRNAs) are often implicated in regulating imprinted expression [16,17,18,19,20]. These features play a significant role in establishing and regulating imprinting mechanisms and also present as targets for the development of molecular based therapies [21, 22].
The disruption of imprinting processes during gametogenesis and the expression of imprinted genes causes significant developmental defects and diseases in humans referred to as genomic imprinting disorders [6, 9, 23]. Epigenome wide association studies and genome wide differentially methylated region analyses have found aberrant epigenetic changes in imprinted and non-imprinted loci. These changes are either germline or somatic origin that could be associated with genetic variants or environmental insults such as nutritional factors and endocrine-disrupting chemicals etc [9]. Somatic origin changes are frequently cell, tissue type, and developmental stage specific [14, 24,25,26]. These changes are referred to as “Epimutations” collectively [9, 27]. The causal role of epimutations in cancer susceptibility has been better characterized over the last two decades but remain to be established whether these changes are implicated in non-cancer related diseases [28,29,30].
Prader–Willi (PWS) and Angelman syndrome (AS) are the first examples of genomic imprinting disorders described in the late 1980s [31,32,33,34]. Currently, there are 15 genomic imprinting disorders described in humans (Table 1). The clinical features reported in imprinting disorders span many organ systems and functional domains and are usually debilitating and lifelong conditions. Neurodevelopmental and neuropsychiatric presentations such as intellectual disability (ID), autism spectrum disorder (ASD), and other psychiatric presentations are notable features associated with the majority of imprinting disorders [35]. For example, psychosis is reported in 10–20% of adult PWS patients and more common in cases resulting from maternal uniparental disomy (UPD) [36].
Recent large scale genomic studies have uncovered a list of genes that encode proteins of epigenetic machinery implicated in neurodevelopmental disorders (NDD) specifically for ASD [37,38,39,40]. For example, allelic specific modifications such as DNA methylation and histones are frequently associated with imprinted genes [41, 42]. Mutations in genes encoding DNA methyltransferase (DNMT3A), DNA demethyltransferase (TET3), H1 linker histone, histone modifying enzymes, and chromatin remodelers (KDM5B, EZH2, EHMT1, CTCF, etc.) have been implicated in ASD and NDD [40, 43,44,45]. It remains to be investigated whether deficiencies of these proteins indirectly affect the genomic imprinting process and their involvement in imprinting disorders.
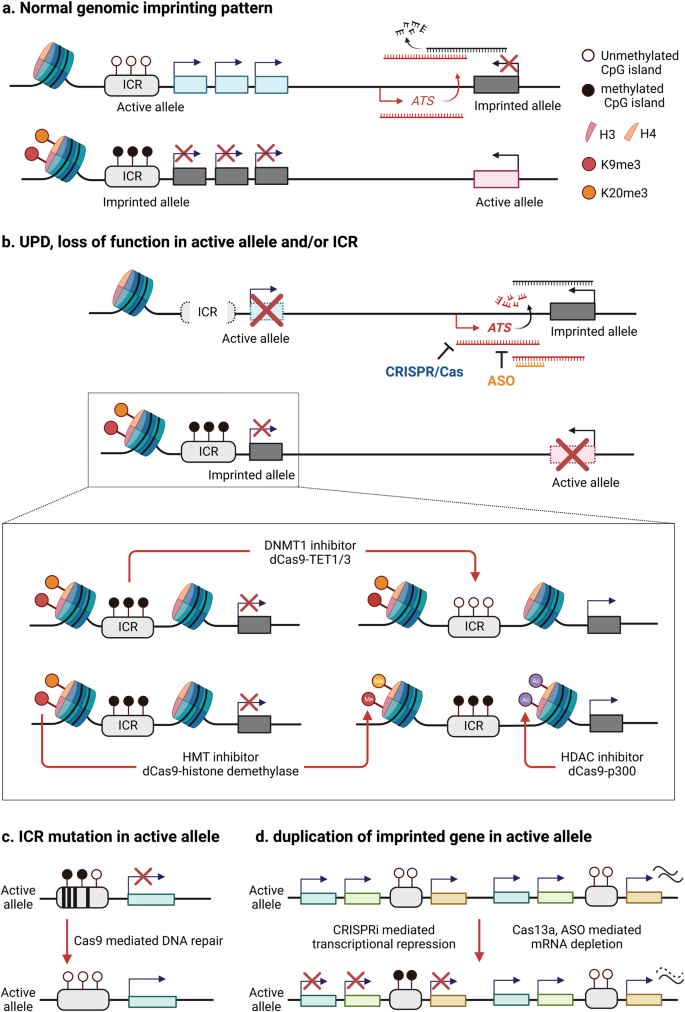
Like most other genetic disorders, there are no effective molecular treatments for imprinting disorders. Current treatments for imprinting disorders are symptom-based interventions and often ineffective. The molecular mechanism underlying imprinting disorders is primarily due to loss of active alleles or duplication of repressed or inactive alleles via different genetic mechanisms [9, 12, 46]. In rare cases, duplication of an active allele can also cause an imprinting disorder [11, 47, 48]. Common genetic defects include a copy number variant (CNV) resulting in loss (e.g. chromosome deletion), DNA sequence variants in an active allele resulting in loss of function of an imprinted gene, and uniparental disomy (UPD) of the repressed allele for an imprinted gene. More rare genetic etiologies include microdeletions or epimutations in the imprinting center/control region (ICR), a regulatory element that controls allele-specific expression of genes in an imprinting domain. Allele-specific epigenetic markers such as DNA methylation and histone modifications are frequently reported in the ICR region [9]. Repressed epigenetic markers are usually associated with repressed alleles and vice versa. These unique molecular features render an exciting opportunity to explore epigenetic-based therapy for imprinting disorders (Fig. 1).
Schematic diagram describes epigenetic-based therapeutic targets including DNA methyltransferase and histone modifying enzymes for imprinting disorders. a Normal genomic imprinting pattern shows parental origin specific allele silencing. b Uniparental disomy (UPD), deletion or mutation of active allele/imprinting control region (ICR) causes deficiency of normal gene expression. CRISPR/dCas9 or small molecule-mediated reactivation of silenced (imprinted) genes is applicable to recover normal gene expression. c Allele specific CRISPR/Cas9-mediated genome editing can be a tool for correction of ICR mutation resulting in an imprinting defect or epimutation. Designing allele specific gRNA is required to do single nucleotide polymorphism (SNP) analysis to distinguish mat/pat chromosome. d Two copies of imprinted gene in active allele can be repressed by CRISPRi (ex. dCas9-KRAB, DNMT1), CRISPR/Cas13 and ASO mediated mRNA depletion (ASO antisense oligonucleotides, ATS antisense transcripts, DNMT1 DNA methyltransferase 1, HMT histone methyltransferase, HDAC Histone deacetylase). Created with BioRender.com.
Epigenetic-based therapy has been investigated extensively in cancers and mostly used the pharmacological approach [49,50,51]. The knowledge learned from cancer studies provides valuable insights for the mechanism and paves a convenient pathway for preclinical imprinting disorder studies in terms of the requirement of regulatory processes for US Food and Drug Administration (FDA) approval. This review describes the exciting development of potential epigenetic therapies targeting imprinting disorders by manipulating chromatin remodeling factors at the level of histone, DNA, and RNA by different molecular approaches. We will highlight CRISPR/Cas9, Cas13 or dCas9 mediated epigenome editing as potential therapies for imprinting disorders using AS and PWS as prime examples.
Scientific premise for epigenetic-based therapy
The molecular basis of epigenetic based therapy for imprinting disorders is to unsilence/reactivate the expression of disease-causing genes from the repressed allele in imprinted loci via pharmacological or molecular genetic manipulation. The basic concept of epigenetic-based therapy has been intensely studied in cancers for more than two decades mostly by pharmacological approach [49,50,51]. Many drugs have been developed or tested that could affect epigenetic modifications at DNA and histone components. Recently, molecular approaches such as antisense oligonucleotide (ASO) [52], shRNA, and CRISPR/Cas9 mediated gene editing have emerged as major interests. The difference between cancers and imprinting disorders is that epigenetic-based therapy for imprinting disorders is allele-specific which is not the case in cancers. Cancer treatments are typically organ or cell-type specific given the nature of somatic mutations but imprinting disorders are mostly of germline origin. In recent years, studies exploring epigenetic-based therapy for imprinting disorders have gained interest and momentum because of the FDA approval of launching several clinical trials using epigenetic-based approach. The basic scientific premise and approach for epigenetic based therapy is diagrammed in Fig. 1 and discussed in detail below. Conceptually, the design of epigenetic-based therapy may target the individual imprinted gene or the imprinting center region that is expected to change the expression of a cluster of imprinted genes within the imprinting domain.
Small molecules for epigenetic modifications
Epigenetic modifications can be achieved by small molecules or other pharmacological interventions altering the function of epigenetic machinery directly associated with transcriptional regulation of imprinted genes. Histone modifying enzymes are common targets for development of novel therapies for human diseases, primarily for cancers. Many small molecules targeting histone deacetylases (HDACs) have been approved by the FDA and are in clinical trials and or used as therapies for cancers and other diseases [53]. These include vorinostat, belinostat, romidepsin, tucidinostat and panobinostat. Significant side effects are reported in previous studies associated with these drugs [54]. Vorinostat is currently in trials for Alzheimer’s disease (NCT03056495) as well as epilepsy (NCT03894826). Valproic acid (VPA, Depakene) is a widely prescribed drug to treat epilepsy in children and used as a mood stabilizer in adults. It has been shown experimentally that VPA possesses HDAC activity, although it is unknown whether this is the mechanism underlying its clinical efficacy. Recent evidence suggests that epigenetic aberrations do play a role in epileptogenesis and that HDAC inhibition may be an attractive mechanism to target for treatment and prevention of epilepsy [55, 56]. Similar to VPA, preclinical studies of Phenylbutyrate (PBA) suggest a possible role as a HDAC inhibitor, and is currently FDA approved for treatment of urea cycle disorders in the form of sodium (Buphenyl) and glycerol (Ravicti) salts. Recently, PBA has been explored in phase 1 and 2 clinical trials for treatment of a range of neurodegenerative diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis [57, 58], and Parkinson’s disease (NCT02046434).
To systemically identify new epigenetic drugs, high content small molecule screenings have been carried out to examine the feasibility of reactivating the repressed imprinted genes using proper markers in cell-based assays [22]. Screenings of small molecule libraries have been performed to investigate potential therapeutic targets for imprinting disorders using mouse models. Using the primary neurons from a reporter mouse model that carries YFP fused to AS Ube3a gene, Philpot and Roth groups have screened >3000 small molecules [59]. This screen identifiies a class of topoisomerase inhibitor that unsilences the expression of AS Ube3a gene from the repressed paternal chromosome in neurons both in vitro and in vivo. One of these topoisomerase inhibitors is topotecan, a FDA approved drug for treatment of metastatic cancers. Intervention with topotecan in the AS Ube3a maternal deficiency mouse model rescues the neurobehavioral phenotypes. The significant toxicity associated with topotecan has precluded it from being considered further in human AS. In a mouse model for PWS, EHMT2/G9a inhibitors are found as effective small molecules to unsilence the expression of normal repressed PWS candidate genes from the maternal chromosome 15q11-q13 region in both human PWS derived cells and PWS mouse model [60]. The biological effect of EHMT2/G9a inhibitors is to reduce the H3K9me2 level. Interestingly, treatment with an EHMT2/G9a inhibitor reduces the level of H3K9me2 in the PWS-ICR without changing its DNA methylation. While the treatment of EHMT2/G9a inhibitor is well tolerated in rodents at different ages, it remains to be investigated whether these EHMT2/G9a inhibitors are safe in humans. In a mouse model for Birk–Barel syndrome, HDAC inhibitor reactivates the paternal silenced Kcnk9 allele with an increase of H3K27 acetylation in Kcnk9 promoter and intronic regions [61]. This inhibitor does not change the allele-specific DNA methylation of Peg13, a differentially methylated region located upstream of Kcnk9 gene [61]. These similar approaches are applicable for identification of effective small molecules to modulate the expression of repressed genes in other imprinting disorders. The biggest challenge of translating these molecules into treatment of human imprinting disorders is the specificity and toxicity of these small molecules. In contrast to drugs targeting terminal cancers using small molecules, the safety threshold of targeting patients with imprinting disorders should be higher.
Antisense oligonucleotide (ASO) mediated reactivation of imprinted genes
ASOs are short, synthetic, single-stranded oligodeoxynucleotides that bind to target pre-mRNAs [62]. Chemically modified ASOs are internalized by active transport or passive diffusion for nucleus entry [63]. Depending on their design and chemical modification, ASOs regulate mRNA levels or alternative splicing through different mechanisms, resulting in changes of mRNA and protein expression [62].
As a therapeutic strategy, ASOs have been introduced and extensively tested for over two decades to improve their activity in clinical trials [64]. With technical advances in their efficacy, about 10 ASO-mediated therapies have been approved by the FDA for genetic and non-genetic disorders [65,66,67]. The most notable success is the ASO treatment of spinal muscular atrophy [68]. These FDA approved ASO based treatments for genetic disorders have paved the way to obtain FDA approval for imprinting disorders. ASO based treatment has emerged as a promising molecular treatment for AS. The AS UBE3A gene is subject to brain specific imprinting [69, 70]. The UBE3A gene is exclusively expressed from the maternal chromosome in neurons [71]. The exact mechanism underlying the brain and neuron specific imprinting for the AS UBE3A gene remains fully characterized. It has been demonstrated in rodent models that the maternal and neuron specific expression of Ube3a is mediated by the expression of a paternally expressed long non-coding antisense RNA to Ube3a (Ube3a-ATS) [18, 72]. Inhibition or inactivation of Ube3a-ATS by ASO and CRISPR based gene editing at the DNA and RNA levels unsilence the expression of sense Ube3a in brains [73,74,75,76]. The reactivation of Ube3a by ASO is capable of rescuing the neurobehavioral and neurophysiological impairments in AS maternal Ube3a deficiency mouse model [73, 77,78,79]. These findings led to a successful approval of investigational new device (IND) for using ASO in treating human AS. In 2020, the FDA approved the first ever phase 1 trial for using ASO via intrathecal injection in AS. Currently, there are 4 active phase 1/2a clinical trials using different ASO designs sponsored by IONIS (NCT05127226), Hoffmann-La Roche (NCT04428281), and Ultragenyx (NCT04259281) in the US and other countries. While the phase 1/2a trials are not primarily designed to assess clinical efficacy, the assessments from these trials revealed encouraging positive signals in multiple behavioral domains [52].
CRISPR/Cas9 genome editing
The studies of ASOs provide a proof of concept to support molecular therapy by manipulating UBE3A-ATS. However, due to the transient nature of ASO treatment after entry into cells, repeated intrathecal injections are necessary to maintain clinical efficacy if it eventually becomes a standard therapy. The requirement of sedation for intrathecal administration of ASOs poses significant medical and psychosocial stress to AS children and families. CRISPR/Cas9 gene editing offers an attractive alternative to ASO. Two recent studies have demonstrated that Cas9 mediated gene editing can inactivate the expression of Ube3a-ATS and reactivate the expression of Ube3a from the paternal chromosome in vitro and in vivo using an adeno-associated virus (AAV) or lentivirus delivery method [74, 75]. A single intrathecal delivery could achieve long term and probably permanent molecular efficacy. Similar to ASOs, the Cas9 editing of Ube3a-ATS rescues the neurobehavioral phenotypes in the AS Ube3a maternal deficiency mouse model. These studies provide initial evidence supporting the feasibility of using Cas9 editing to treat AS. Like CRISPR/Cas9 mediated gene editing in other genetic diseases, safety concerns related to virus delivery methods and potential off-target effects due to Cas9 editing remain to be evaluated thoroughly before moving to human trials. Conceptually, an unbiased CRISPR based screening could be designed to screen for a genetic locus that could unsilence imprinted genes and lead to the development of a new treatment. This has not been reported in literature so far.
CRISPR mediated RNA editing
Non-coding RNAs are frequently associated with imprinted gene clusters and strongly implicated in the regulation of imprinted clusters [16, 80,81,82]. The CRISPR/Cas13 system is an effective tool for RNA editing [83], where Cas13 controls RNA without permanent change of DNA sequence in gene bodies. In contrast, Cas9 has technical limitations including low editing efficiency, higher probability of inducing off-target events and oversized AAV packaging [84]. Cas13 modulates RNA readout through various modifications such as methylation, demethylation, and A-I/C-U editing. Cas13a with gRNA has proven RNA-guided RNA knockdown [85,86,87]. Conceptually, this tool can be used to target specific lncRNAs controlling imprinted domains or reactivate imprinted genes. dCas13b-ADAR2 has a function of A to I RNA base editing [83, 84], which has the important role of correcting pathogenic mutations at the RNA level. dCas13-METTL3/14 as a N6-adenosine-methyltransferase (A to m6A), increases RNA stability and translation efficiency, leading to enhancement of gene expression [84]. CRISPR-Cas13d variants such as dCasRx is the smallest form targeting RNA, and is able to be packaged into lentivirus for improved delivery efficiency to primary cells [88]. As a m6A demethylase, dCasRx-ALKBH5 shows bidirectional modulation depending on targeting mRNA [89]. The technical challenge for the translational potential of Cas13 may be related to targeting deliverance of the editing tool to specified organs and cell types. Recently, the use of Cas13 based editing of UBE3A-ATS has been reported to reactivate the expression of UBE3A from the paternal chromosome and rescue some neurobehaviors in mice [76]. However, like ASOs, the transient effect of Cas13 RNA editing is expected to require repeated interventions to maintain efficacy.
CRISPR/dCas9 mediated epigenome editing
CRISPR-based epigenome editing technologies have been developed to enable manipulation of the epigenome and regulate expression of targeted genes [90,91,92,93,94]. For the design of epigenome editing, a catalytically inactive mutant form of Cas9 (dCas9) without endonuclease activity still binds to target DNA sequence that matches guide RNA. dCas9 fused with an effector domain has emerged as a popular approach to target a specific locus with specific modifications. The fusion constructs of dCas9 include various catalytic domains of epigenetic modifying enzymes and chromatin remodelers that have been demonstrated to result in transcriptional activation or repression for a targeted gene. Recently, Cas13 has been shown to improve the dCas9 platform targeting DNA and histones or recruiting other transcriptional factors [83, 86]. Chemically modified guide RNA could also improve the transcript knockdown efficiency with CRISPR/Cas13 as a form of ribonucleoprotein (RNP) complex [76, 95]. Conceptually, epigenome editing is applicable to target allele-specific DNA and histone modifications associated with repressed alleles and reactivate the expression of imprinted genes [96].
DNA methylation and demethylation modification and editing for imprinted genes
5-methylcytosine (5mC) in CG dinucleotides (CpG) plays a critical role in imprinting establishment during development [97,98,99]. CpG islands are mainly located in transcriptional regulatory elements such as promoter, inhibitor, and enhancer regions [100]. In the imprinting domain, allelic methylation of CpG dinucleotides is frequently identified in the imprinting center. DNA methyltransferase (DNMT), an epigenetic writer, has a primary role in establishing and maintaining DNA methylation, resulting in subsequent recruitment of repressor complex for gene silencing in general. dCas9 fused with mammalian DNA methyltransferase of DNMT1, DNMT3A, DNMT3B, and DNMT3L and prokaryotic DNA methyltransferase MQ1 displayed de novo methylation with inhibition of transcription in preclinical studies [101,102,103,104,105]. This tool can be applied to imprinting disorders caused by duplication of active allele, such as transient neonatal diabetes mellitus type 1 (paternal duplication of 6q24) and Beckwith–Wiedemann syndrome (paternal duplication of 11q15.5) [106].
As an enzymatic eraser, the ten-eleven translocation dioxygenase family of genes (TET1-3) encodes enzymes that oxidize 5mC to 5-hydroxymethylcytosine (5hmC) leading to active DNA demethylation and increase of gene transcription in general. dCas9-TET1/TET3 targeting to methylated CpG island contributes to specific gene activation in various disease models [92, 107,108,109]. The dCas9-TET1 fusion construct can demethylate the methylated 5mC associated CGG repeat expansion of FMR1 (Fragile X Syndrome) in cells and in vivo [102], the CpG island of the maternally imprinted Snrpn gene in a rodent model [92], and the MECP2 promoter for its reactivation from inactivated Xi chromosome in Rett syndrome human embryonic stem cells and derived neurons [110]. With the same principle, the dCas9-TET fusion protein can be used to reactivate imprinted genes that have been shown to be associated with methylated CpG islands in regulatory regions.
Histone modification editing
Manipulating posttranslational modifications of histones in in vitro and in vivo experimental systems using CRISPR/dCas9-mediate editing have shown a strong effect on gene regulation bidirectionally as well as specificity. For example, dCas9-p300 and dCas9-dMSK1 induce an increase of target gene transcription through acetylation and phosphorylation to specific residues on histones, respectively [111, 112] (Table 2). Conversely, dCas9-HDAC1/3 silences gene expression by deacetylation of histone residues to induce compact chromatin status [113, 114]. Activity of dCas9 fused with histone methyltransferase or demethylase is dependent on cell type and developmental stages [115, 116]. As summarized in Table 2, dCas9-histone methyltransferase usually leads to gene silencing with other repressor components. A few of those enzymes show bi-directional regulations that depend on gene loci and associated molecular context [117, 118]. Thus, it is necessary to understand specific mechanisms in tissues or cell lines that are associated with imprinted impression when designing translational applications.
To minimize off-target events and enhance precision, an inducible transient expression system has been introduced for epigenome editing [119,120,121]. Inducible dCas9 expression or activation allows tracking of its spatiotemporal control and potentially minimize off-target events because of the transient expression of dCas9. Technically, dCas9 fusion proteins are limited by their sizes over the maximal cargo size of AAV, a common and popular delivery tool. Novel delivery tools are necessary for in vivo application and clinical trials in humans. Alternative non-viral tools such as RNP or nanoparticle mediated delivery have emerged as better platforms, which have no limitations of package size [122, 123]. However, the molecular weight of current RNP or nanoparticle designs are too big to penetrate the brain efficiently. The cell type specificity of RNP and nanoparticle mediated delivery is poorly understood and this may limit clinical applications.
Multiplexed CRISPRi and CRISPRa system
CRISPR/dCas9 mediated transcriptional interference (CRISPRi) or transcriptional activation (CRISPRa) systems have been actively developed due to eagerness for efficacy improvement. Various advanced versions of these systems can recruit multiple transcription factors to control gene transcription. More than two kinds of catalytic domains are fused with dCas9 for synergetic effects on target gene regulation [91, 96]. Beyond the introduction of four copies of herpes simplex viral protein 16 (VP64) as an activator, fusion or recruitment of multiple transcriptional factors have shown improved efficiency when manipulating target gene loci [124, 125] (Table 2). With dCas9 development, there have been trials for using combined or multiple gRNAs for more dramatic potency in target gene expression [126].
For epigenetic gene silencing, dCas9 is fused with Krüppel associated box (KRAB), derived from zinc finger domain, leading to decrease of chromatin accessibility and high H3K9me3 levels on target regulatory regions with recruitment of other repressors [127]. An advanced version of CRISPRi, dCas9-KRAB-MeCP2, has been shown to be highly effective as a transcriptional repressor [128]. While most dCas9 fusion proteins have cis-regulatory features, dCas9-HP1α acts in cis and trans by tethering to reach distal regulatory elements [129]. This tool also reduces chromatin accessibility by transcription machineries maintaining compact chromatin status.
Illustrated examples of epigenetic therapy strategies for AS and PWS
AS and PWS are caused by deficiency of maternally and paternally expressed genes on chromosome 15q11-q13, respectively. De novo ~6 Mb paternal deletion of the 15q11-q13 region is found in ~70% of PWS patients, followed by maternal UPD (~27%), and rare imprinting center mutations (Table 3) [130]. There are a dozen paternally expressed genes within the 15q11-q13 region (Fig. 2). SNORD116 is considered to be the critical gene, where a deficiency is responsible for key PWS clinical features [21, 131]. The allele-specific expression of paternally expressed genes in the 15q11-q13 region is controlled by an imprinting center (PWS-IC) located upstream of the SNURF-SNRPN gene [132, 133]. The CpG islands located in the PWS-IC are unmethylated in the paternal chromosome but methylated in the maternal chromosome. Allele-specific histone modifications such as H3K9me2 are also associated with PWS-IC [60, 134]. These unique epigenetic defects render an opportunity to explore chromatin remodeling enzymes as therapeutic targets [21, 22]. Treatment with a DNA methylation inhibitor can reactivate the expression of SNRPN from the maternal chromosome in cells derived from a patient with PWS due to a paternal deletion of 15q11-q13 [135, 136]. In preclinical studies, treatment of EHMT2/G9a inhibitors have shown promise as potential pharmacological small molecules by reducing H3K9me2 levels, leading to reactivation of the imprinted gene without change of DNA methylation in a PWS mouse model [60]. Similarly, a fusion construct of dCas9 with H3K9me2/3 demethylase may be able to reduce the histone H3K9 methylation level at PWS-IC, resulting in reactivation of imprinted genes on the maternal chromosome. dCas9-TET1 could demethylate the Snrpn promoter region including CpG islands, suggesting a potent alternative tool [92]. Further preclinical studies are warranted to explore the therapeutic potential in human PWS.

Schematic shows the imprinting domain in human chromosome 15q11-q13 with potential epigenetic therapeutic candidates for (a) AS and (b) PWS. Genes in dark blue are exclusively expressed from the paternal chromosome while genes in purple are expressed from the maternal chromosome in neuronal cell type specific manner (gray bar, imprinted gene; biallelic expressed gene, black bar). In the case of AS, the loss of UBE3A expression in the maternal allele by different mechanisms is the cause. The principle of epigenetics-based therapy is to reactivate the paternal allele’s expression of UBE3A in neurons. The current approach is to inhibit the expression of antisense of UBE3A via small molecule, ASO, CRISPR/Cas9, or Cas13. In the case of PWS, where more than one paternally expressed gene is in the candidate region, the optimal approach is to manipulate the imprinting center region to reactivate the expression of silenced genes from the maternal chromosome. Current approaches include DNA methylation inhibitor, small molecule for histone modifications and others, CRISPR/dCas9 gene editing. Created with BioRender.com.
In the case of AS, most patients (70%) have a de novo ~6 Mb maternal deletion including the UBE3A gene. About 10% have mutations within the UBE3A gene, followed by paternal uniparental disomy of chromosome 15 and imprinting center mutations [137,138,139]. UBE3A is the only maternally expressed gene within the chromosome 15q11-q13 region [69]. The expression of the maternal UBE3A allele is brain or neuron specific (Table 3) [70, 71, 140]. Intriguingly, there is no allele-specific epigenetic modification associated with AS-IC. The repressed expression of Ube3a in the paternal chromosome is mediated by a paternally expressed long non-coding RNA from the upstream region of Snrpn [18, 72]. The development of epigenetic and molecular therapy for AS has significantly advanced over the last decade [141]. Topotecan was the first drug showing a robust reactivation of Ube3a in a preclinical study in AS mouse model [59]. However, its translational potential is limited because of significant toxicity. The development of a safer new class of topoisomerase inhibitors remains a promising molecular therapy for AS. Treatment with topotecan can reduce the expression of UBE3A-ATS and reactivate the expression of UBE3A in human derived neurons and mouse neurons [59, 142]. Both ASO and Cas9 editing of Ube3a-ATS unsilence the paternal Ube3a allele by reducing Ube3a-ATS in AS mouse model [73,74,75, 143]. The success of ongoing ASO clinical trials will certainly promote translational studies exploring the use of Cas9 editing in human AS. The application of CRISPR/Cas13 mediated gRNA targeting UBE3A-ATS has been shown to be effective in AS mouse model [76]. Additional studies are warranted before advancing to IND studies.
Concluding remark: the promises and challenges
With the FDA approved ASO in phase 1/2a trial for AS, the prospect of developing epigenetic molecular therapies for other genomic imprinting disorders is encouraging. The number of genomic imprinting disorders and the populations affected are relatively small. However, because of the unique molecular defects associated with genomic imprinting disorders and molecular mechanisms underlying imprinting regulations, genomic imprinting disorders remain the best opportunity for a proof of principle study of developing epigenetic therapy for genetic diseases. The lessons learned and tools developed are immediately applicable to other non-imprinting genetic disorders. For example, genetic defects that lead to haploinsufficiency of single genes are found in 10–15% of cases with ASD and even higher in other NDD. The approach of upregulating the expression of normal alleles by epigenome editing is an attractive avenue for developing molecular treatments. Despite these promises, challenges remain significant. For pharmacological based epigenetic therapy, the broad effects of epigenetic modulating drugs remain a potential concern for clinical applications particularly for mild or moderate presentation of the disease. It is possible that side effects are dose dependent and likely cell and tissue type specific. The careful assessment of these issues may ease the concern from FDA regulatory concerns. For ASO and gene editing based therapies, the efficiency of delivery platforms remains to be improved especially for the brain as a target organ. For example, the repeat dosing for ASO intrathecal delivery used in AS clinical trials is not optimal for patient care. The virus delivery platform for Cas9 editing has its own inherited and well recognized limitations. New non-viral delivery platforms have been developed in recent years but they are suboptimal for brain disorders. Lastly, for any gene editing approach, the genome wide off-target events of Cas9/gRNA remain a concern conceptually. The challenge is to assess the impact of off-target events technically and physiologically, and how to define an acceptable threshold of genome wide off-target events from a regulatory and safety perspective. Nevertheless, it is reasonable to predict that more promising breakthroughs utilizing CRISPR based genome and epigenome editing will emerge as an effective treatment modality in the near future.
References
-
Surani MA, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 1984;308:548–50.
Google Scholar
-
McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 1984;37:179–83.
Google Scholar
-
Barlow DP, Stoger R, Herrmann BG, Saito K, Schweifer N. The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature. 1991;349:84–7.
Google Scholar
-
Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014;141:1805–13.
Google Scholar
-
Perez JD, Rubinstein ND, Dulac C. New perspectives on genomic imprinting, an essential and multifaceted mode of epigenetic control in the developing and adult brain. Annu Rev Neurosci. 2016;39:347–84.
Google Scholar
-
Nicholls RD. The impact of genomic imprinting for neurobehavioral and developmental disorders. J Clin Invest. 2000;105:413–8.
Google Scholar
-
Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3:7.
Google Scholar
-
Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 2014;6:2.
Google Scholar
-
Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. 2019;20:235–48.
Google Scholar
-
Lucifero D, Chaillet JR, Trasler JM. Potential significance of genomic imprinting defects for reproduction and assisted reproductive technology. Hum Reprod Update. 2004;10:3–18.
Google Scholar
-
Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tumer Z, Monk D, et al. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics. 2015;7:123.
Google Scholar
-
Horsthemke B, Zechner U. Novel strategies to cure imprinting disorders. Medizinische Genetik. 2020;32:335–40.
Google Scholar
-
Lleres D, Moindrot B, Pathak R, Piras V, Matelot M, Pignard B, et al. CTCF modulates allele-specific sub-TAD organization and imprinted gene activity at the mouse Dlk1-Dio3 and Igf2-H19 domains. Genome Biol. 2019;20:272.
Google Scholar
-
Raas MWD, Zijlmans DW, Vermeulen M, Marks H. There is another: H3K27me3-mediated genomic imprinting. Trends Genet. 2022;38:82–96.
Google Scholar
-
Monnier P, Martinet C, Pontis J, Stancheva I, Ait-Si-Ali S, Dandolo L. H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc Natl Acad Sci USA. 2013;110:20693–8.
Google Scholar
-
Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002;415:810–3.
Google Scholar
-
Hayward BE, Bonthron DT. An imprinted antisense transcript at the human GNAS1 locus. Hum Mol Genet. 2000;9:835–41.
Google Scholar
-
Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 2013;9:e1004039.
Google Scholar
-
Williamson CM, Ball ST, Dawson C, Mehta S, Beechey CV, Fray M, et al. Uncoupling antisense-mediated silencing and DNA methylation in the imprinted Gnas cluster. PLoS Genet. 2011;7:e1001347.
Google Scholar
-
MacDonald WA, Mann MRW. Long noncoding RNA functionality in imprinted domain regulation. PLoS Genet. 2020;16:e1008930.
Google Scholar
-
Kim Y, Wang SE, Jiang YH. Epigenetic therapy of Prader-Willi syndrome. Transl Res. 2019;208:105–18.
Google Scholar
-
Wang SE, Jiang YH. Potential of epigenetic therapy for Prader-Willi syndrome. Trends Pharm Sci. 2019;40:605–8.
Google Scholar
-
Falls JG, Pulford DJ, Wylie AA, Jirtle RL. Genomic imprinting: implications for human disease. Am J Pathol. 1999;154:635–47.
Google Scholar
-
Li Y, Li J. Technical advances contribute to the study of genomic imprinting. PLoS Genet. 2019;15:e1008151.
Google Scholar
-
Kupers LK, Monnereau C, Sharp GC, Yousefi P, Salas LA, Ghantous A, et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat Commun. 2019;10:1893.
Google Scholar
-
Docherty LE, Rezwan FI, Poole RL, Jagoe H, Lake H, Lockett GA, et al. Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J Med Genet. 2014;51:229–38.
Google Scholar
-
Oey H, Whitelaw E. On the meaning of the word ‘epimutation’. Trends Genet. 2014;30:519–20.
Google Scholar
-
Goovaerts T, Steyaert S, Vandenbussche CA, Galle J, Thas O, Van Criekinge W, et al. A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat Commun. 2018;9:4120.
Google Scholar
-
Kim J, Bretz CL, Lee S. Epigenetic instability of imprinted genes in human cancers. Nucleic Acids Res. 2015;43:10689–99.
Google Scholar
-
Holm TM, Jackson-Grusby L, Brambrink T, Yamada Y, Rideout WM III, Jaenisch R. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell. 2005;8:275–85.
Google Scholar
-
Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. 1989;342:281–5.
Google Scholar
-
Jiang Y, Tsai TF, Bressler J, Beaudet AL. Imprinting in Angelman and Prader-Willi syndromes. Curr Opin Genet Dev. 1998;8:334–42.
Google Scholar
-
Butler MG, Meaney FJ, Palmer CG. Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet. 1986;23:793–809.
Google Scholar
-
Knoll JH, Nicholls RD, Magenis RE, Glatt K, Graham JM Jr, Kaplan L, et al. Angelman syndrome: three molecular classes identified with chromosome 15q11q13-specific DNA markers. Am J Hum Genet. 1990;47:149–54.
Google Scholar
-
Isles AR. The contribution of imprinted genes to neurodevelopmental and neuropsychiatric disorders. Transl Psychiatry. 2022;12:210.
Google Scholar
-
Vogels A, De Hert M, Descheemaeker MJ, Govers V, Devriendt K, Legius E, et al. Psychotic disorders in Prader-Willi syndrome. Am J Med Genet A. 2004;127A:238–43.
Google Scholar
-
Zhou X, Feliciano P, Shu C, Wang T, Astrovskaya I, Hall JB, et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet. 2022;54:1305–19.
Google Scholar
-
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180:568–84.e523.
Google Scholar
-
Fu JM, Satterstrom FK, Peng M, Brand H, Collins RL, Dong S, et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet. 2022;54:1320–31.
Google Scholar
-
Duffney LJ, Valdez P, Tremblay MW, Cao X, Montgomery S, McConkie-Rosell A, et al. Epigenetics and autism spectrum disorder: a report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am J Med Genet B Neuropsychiatr Genet. 2018;177:426–33.
Google Scholar
-
Hanna CW, Kelsey G. Genomic imprinting beyond DNA methylation: a role for maternal histones. Genome Biol. 2017;18:177.
Google Scholar
-
Chen Z, Yin Q, Inoue A, Zhang C, Zhang Y. Allelic H3K27me3 to allelic DNA methylation switch maintains noncanonical imprinting in extraembryonic cells. Sci Adv. 2019;5:eaay7246.
Google Scholar
-
Polonis K, Blackburn PR, Urrutia RA, Lomberk GA, Kruisselbrink T, Cousin MA, et al. Co-occurrence of a maternally inherited DNMT3A duplication and a paternally inherited pathogenic variant in EZH2 in a child with growth retardation and severe short stature: atypical Weaver syndrome or evidence of a DNMT3A dosage effect? Cold Spring Harb Mol Case Stud. 2018;4:4.
Google Scholar
-
Mossink B, Negwer M, Schubert D, Nadif, Kasri N. The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell Mol Life Sci. 2021;78:2517–63.
Google Scholar
-
Beck DB, Petracovici A, He C, Moore HW, Louie RJ, Ansar M, et al. Delineation of a human mendelian disorder of the DNA demethylation machinery: TET3 deficiency. Am J Hum Genet. 2020;106:234–45.
Google Scholar
-
Butler MG, Duis J. Chromosome 15 imprinting disorders: genetic laboratory methodology and approaches. Front Pediatr. 2020;8:154.
Google Scholar
-
Horsthemke B. In brief: genomic imprinting and imprinting diseases. J Pathol. 2014;232:485–7.
Google Scholar
-
Monk D, Wakeling EL, Proud V, Hitchins M, Abu-Amero SN, Stanier P, et al. Duplication of 7p11.2-p13, including GRB10, in Silver-Russell syndrome. Am J Hum Genet. 2000;66:36–46.
Google Scholar
-
Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Annu Rev Med. 2016;67:73–89.
Google Scholar
-
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Google Scholar
-
Bates SE. Epigenetic therapies for cancer. N Engl J Med. 2020;383:650–63.
Google Scholar
-
Tjeertes J, Bacino CA, Bichell TJ, Bird LM, Bustamante M, Crean R, et al. Enabling endpoint development for interventional clinical trials in individuals with Angelman syndrome: a prospective, longitudinal, observational clinical study (FREESIAS). J Neurodev Disord. 2023;15:22.
-
Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Schioth HB. Recent developments of HDAC inhibitors: emerging indications and novel molecules. Br J Clin Pharm. 2021;87:4577–97.
Google Scholar
-
Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92.
-
Reddy SD, Clossen BL, Reddy DS. Epigenetic histone deacetylation inhibition prevents the development and persistence of temporal lobe epilepsy. J Pharm Exp Ther. 2018;364:97–109.
Google Scholar
-
Romoli M, Mazzocchetti P, D‘Alonzo R, Siliquini S, Rinaldi VE, Verrotti A, et al. Valproic acid and epilepsy: from molecular mechanisms to clinical evidences. Curr Neuropharmacol. 2019;17:926–46.
Google Scholar
-
Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383:919–30.
Google Scholar
-
Gauthier S, Alam J, Fillit H, Iwatsubo T, Liu-Seifert H, Sabbagh M, et al. Combination therapy for Alzheimer’s disease: perspectives of the EU/US CTAD task force. J Prev Alzheimers Dis. 2019;6:164–8.
Google Scholar
-
Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481:185–9.
Google Scholar
-
Kim Y, Lee HM, Xiong Y, Sciaky N, Hulbert SW, Cao X, et al. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat Med. 2017;23:213–22.
Google Scholar
-
Cooper A, Butto T, Hammer N, Jagannath S, Fend-Guella DL, Akhtar J, et al. Inhibition of histone deacetylation rescues phenotype in a mouse model of Birk-Barel intellectual disability syndrome. Nat Commun. 2020;11:480.
Google Scholar
-
Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14:9–21.
Google Scholar
-
Lorenz P, Misteli T, Baker BF, Bennett CF, Spector DL. Nucleocytoplasmic shuttling: a novel in vivo property of antisense phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 2000;28:582–92.
Google Scholar
-
Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, et al. Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med. 2020;9:6.
Google Scholar
-
Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19:673–94.
Google Scholar
-
Stein CA, Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol Ther. 2017;25:1069–75.
Google Scholar
-
Hill SF, Meisler MH. Antisense oligonucleotide therapy for neurodevelopmental disorders. Dev Neurosci. 2021;43:247–52.
Google Scholar
-
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:3017–26.
Google Scholar
-
Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–8.
Google Scholar
-
Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–5.
Google Scholar
-
Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, et al. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–47.
Google Scholar
-
Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet. 2012;21:3001–12.
Google Scholar
-
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–12.
Google Scholar
-
Wolter JM, Mao H, Fragola G, Simon JM, Krantz JL, Bazick HO, et al. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature. 2020;587:281–4.
Google Scholar
-
Schmid RS, Deng X, Panikker P, Msackyi M, Breton C, Wilson JM. CRISPR/Cas9 directed to the Ube3a antisense transcript improves Angelman syndrome phenotype in mice. J Clin Invest. 2021;131:e142574.
-
Li J, Shen Z, Liu Y, Yan Z, Liu Y, Lin X, et al. A high-fidelity RNA-targeting Cas13 restores paternal Ube3a expression and improves motor functions in Angelman syndrome mice. Mol Ther. 2023;5;31:2286–95.
-
Copping NA, McTighe SM, Fink KD, Silverman JL. Emerging gene and small molecule therapies for the neurodevelopmental disorder Angelman syndrome. Neurotherapeutics. 2021;18:1535–47.
Google Scholar
-
Davidson BL, Gao G, Berry-Kravis E, Bradbury AM, Bonnemann C, Buxbaum JD, et al. Gene-based therapeutics for rare genetic neurodevelopmental psychiatric disorders. Mol Ther. 2022;30:2416–28.
Google Scholar
-
Milazzo C, Mientjes EJ, Wallaard I, Rasmussen SV, Erichsen KD, Kakunuri T, et al. Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight. 2021;6:15.
Google Scholar
-
Zhang M, Zhao H, Xie S, Chen J, Xu Y, Wang K, et al. Extensive, clustered parental imprinting of protein-coding and noncoding RNAs in developing maize endosperm. Proc Natl Acad Sci USA. 2011;108:20042–7.
Google Scholar
-
Koerner MV, Pauler FM, Huang R, Barlow DP. The function of non-coding RNAs in genomic imprinting. Development. 2009;136:1771–83.
Google Scholar
-
Andergassen D, Muckenhuber M, Bammer PC, Kulinski TM, Theussl HC, Shimizu T, et al. The Airn lncRNA does not require any DNA elements within its locus to silence distant imprinted genes. PLoS Genet. 2019;15:e1008268.
Google Scholar
-
Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, et al. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–27.
Google Scholar
-
Tang T, Han Y, Wang Y, Huang H, Qian P. Programmable system of Cas13-mediated RNA modification and its biological and biomedical applications. Front Cell Dev Biol. 2021;9:677587.
Google Scholar
-
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–4.
Google Scholar
-
Xu C, Zhou Y, Xiao Q, He B, Geng G, Wang Z, et al. Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat Methods. 2021;18:499–506.
Google Scholar
-
Kushawah G, Hernandez-Huertas L, Abugattas-Nunez Del Prado J, Martinez-Morales JR, DeVore ML, Hassan H, et al. CRISPR-Cas13d induces efficient mRNA knockdown in animal embryos. Dev Cell. 2020;54:805–7.e807.
Google Scholar
-
Lo N, Xu X, Soares F, He HH. The basis and promise of programmable RNA editing and modification. Front Genet. 2022;13:834413.
Google Scholar
-
Xia Z, Tang M, Ma J, Zhang H, Gimple RC, Prager BC, et al. Epitranscriptomic editing of the RNA N6-methyladenosine modification by dCasRx conjugated methyltransferase and demethylase. Nucleic Acids Res. 2021;49:7361–74.
Google Scholar
-
Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9:1911.
Google Scholar
-
Nunez JK, Chen J, Pommier GC, Cogan JZ, Replogle JM, Adriaens C, et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184:2503–19.e2517.
Google Scholar
-
Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–47.e217.
Google Scholar
-
Matharu N, Rattanasopha S, Tamura S, Maliskova L, Wang Y, Bernard A, et al. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science. 2019;363:6424.
Google Scholar
-
Kojima S, Shiochi N, Sato K, Yamaura M, Ito T, Yamamura N, et al. Epigenome editing reveals core DNA methylation for imprinting control in the Dlk1-Dio3 imprinted domain. Nucleic Acids Res. 2022;50:5080–94.
Google Scholar
-
Mendez-Mancilla A, Wessels HH, Legut M, Kadina A, Mabuchi M, Walker J, et al. Chemically modified guide RNAs enhance CRISPR-Cas13 knockdown in human cells. Cell Chem Biol. 2022;29:321–7.e324.
Google Scholar
-
Syding LA, Nickl P, Kasparek P, Sedlacek R. CRISPR/Cas9 epigenome editing potential for rare imprinting diseases: a review. Cells. 2020;9:4.
Google Scholar
-
Wang SE, Jiang Y-H. Epigenetic epidemiology of autism and other neurodevelopmental disorders. In: Michels KB, editor. Epigenetic Epidemiology. Cham: Springer International Publishing; 2022. p. 405–26.
-
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38.
Google Scholar
-
Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607.
Google Scholar
-
Cain JA, Montibus B, Oakey RJ. Intragenic CpG islands and their impact on gene regulation. Front Cell Dev Biol. 2022;10:832348.
Google Scholar
-
Pflueger C, Tan D, Swain T, Nguyen T, Pflueger J, Nefzger C, et al. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018;28:1193–206.
Google Scholar
-
Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, et al. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172:979–92.e976.
Google Scholar
-
O’Geen H, Tomkova M, Combs JA, Tilley EK, Segal DJ. Determinants of heritable gene silencing for KRAB-dCas9+DNMT3 and Ezh2-dCas9+DNMT3 hit-and-run epigenome editing. Nucleic Acids Res. 2022;50:3239–53.
Google Scholar
-
Lu A, Wang J, Sun W, Huang W, Cai Z, Zhao G, et al. Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation. Cell Discov. 2019;5:22.
Google Scholar
-
Lei Y, Zhang X, Su J, Jeong M, Gundry MC, Huang YH, et al. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat Commun. 2017;8:16026.
Google Scholar
-
Carli D, Riberi E, Ferrero GB, Mussa A. Syndromic disorders caused by disturbed human imprinting. J Clin Res Pediatr Endocrinol. 2020;12:1–16.
Google Scholar
-
Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7:46545–56.
Google Scholar
-
Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016;34:1060–5.
Google Scholar
-
Xu X, Tan X, Tampe B, Wilhelmi T, Hulshoff MS, Saito S, et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat Commun. 2018;9:3509.
Google Scholar
-
Qian J, Guan X, Xie B, Xu C, Niu J, Tang X, et al. Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Sci Transl Med. 2023;15:eadd4666.
Google Scholar
-
Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–7.
Google Scholar
-
Li J, Mahata B, Escobar M, Goell J, Wang K, Khemka P, et al. Programmable human histone phosphorylation and gene activation using a CRISPR/Cas9-based chromatin kinase. Nat Commun. 2021;12:896.
Google Scholar
-
Liu J, Sun M, Cho KB, Gao X, Guo B. A CRISPR-Cas9 repressor for epigenetic silencing of KRAS. Pharm Res. 2021;164:105304.
Google Scholar
-
Kwon DY, Zhao YT, Lamonica JM, Zhou Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat Commun. 2017;8:15315.
Google Scholar
-
O’Geen H, Bates SL, Carter SS, Nisson KA, Halmai J, Fink KD, et al. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin. 2019;12:26.
Google Scholar
-
Baumann V, Wiesbeck M, Breunig CT, Braun JM, Koferle A, Ninkovic J, et al. Targeted removal of epigenetic barriers during transcriptional reprogramming. Nat Commun. 2019;10:2119.
Google Scholar
-
Lu Z, Yang S, Yuan X, Shi Y, Ouyang L, Jiang S, et al. CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillus subtilis. Nucleic Acids Res. 2019;47:e40.
Google Scholar
-
Savell KE, Bach SV, Zipperly ME, Revanna JS, Goska NA, Tuscher JJ, et al. A neuron-optimized CRISPR/dCas9 activation system for robust and specific gene regulation. eNeuro. 2019;6:1.
Google Scholar
-
Kelkar A, Zhu Y, Groth T, Stolfa G, Stablewski AB, Singhi N, et al. Doxycycline-dependent self-inactivation of CRISPR-Cas9 to temporally regulate on- and off-target editing. Mol Ther. 2020;28:29–41.
Google Scholar
-
Holoch D, Wassef M, Lovkvist C, Zielinski D, Aflaki S, Lombard B, et al. A cis-acting mechanism mediates transcriptional memory at Polycomb target genes in mammals. Nat Genet. 2021;53:1686–97.
Google Scholar
-
Cao J, Wu L, Zhang SM, Lu M, Cheung WK, Cai W, et al. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44:e149.
Google Scholar
-
Chen J, Guo Z, Tian H, Chen X. Production and clinical development of nanoparticles for gene delivery. Mol Ther Methods Clin Dev. 2016;3:16023.
Google Scholar
-
Duan L, Ouyang K, Xu X, Xu L, Wen C, Zhou X, et al. Nanoparticle delivery of CRISPR/Cas9 for genome editing. Front Genet. 2021;12:673286.
Google Scholar
-
Pandelakis M, Delgado E, Ebrahimkhani MR. CRISPR-based synthetic transcription factors in vivo: the future of therapeutic cellular programming. Cell Syst. 2020;10:1–14.
Google Scholar
-
Riedmayr LM, Hinrichsmeyer KS, Karguth N, Bohm S, Splith V, Michalakis S, et al. dCas9-VPR-mediated transcriptional activation of functionally equivalent genes for gene therapy. Nat Protoc. 2022;17:781–18.
Google Scholar
-
Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther. 2020;5:1.
Google Scholar
-
Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12:1143–9.
Google Scholar
-
Yeo NC, Chavez A, Lance-Byrne A, Chan Y, Menn D, Milanova D, et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods. 2018;15:611–6.
Google Scholar
-
Gao Y, Han M, Shang S, Wang H, Qi LS. Interrogation of the dynamic properties of higher-order heterochromatin using CRISPR-dCas9. Mol Cell. 2021;81:4287–99.e4285.
Google Scholar
-
Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle; 1993.
-
Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–21.
Google Scholar
-
Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:365–76.
Google Scholar
-
Sutcliffe JS, Nakao M, Christian S, Orstavik KH, Tommerup N, Ledbetter DH, et al. Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat Genet. 1994;8:52–8.
Google Scholar
-
Xin Z, Tachibana M, Guggiari M, Heard E, Shinkai Y, Wagstaff J. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J Biol Chem. 2003;278:14996–5000.
Google Scholar
-
Fulmer-Smentek SB, Francke U. Association of acetylated histones with paternally expressed genes in the Prader–Willi deletion region. Hum Mol Genet. 2001;10:645–52.
Google Scholar
-
Saitoh S, Wada T. Parent-of-origin specific histone acetylation and reactivation of a key imprinted gene locus in Prader-Willi syndrome. Am J Hum Genet. 2000;66:1958–62.
Google Scholar
-
Bossuyt SNV, Punt AM, de Graaf IJ, van den Burg J, Williams MG, Heussler H, et al. Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum Mol Genet. 2021;30:430–42.
Google Scholar
-
Dagli AI, Mathews J, Williams CA. Angelman syndrome. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al. editors. GeneReviews((R)). Seattle (WA): University of Washington; 1993.
-
Beaudet AL. Genetics of Angelman syndrome. Hum Mol Genet. 1999;65:1–6.
-
Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17:12–3.
Google Scholar
-
Beaudet AL, Meng L. Gene-targeting pharmaceuticals for single-gene disorders. Hum Mol Genet. 2016;25:R18–26.
Google Scholar
-
Sen D, Voulgaropoulos A, Drobna Z, Keung AJ. Human cerebral organoids reveal early spatiotemporal dynamics and pharmacological responses of UBE3A. Stem Cell Rep. 2020;15:845–54.
Google Scholar
-
Dindot SV, Christian S, Murphy WJ, Berent A, Panagoulias J, Schlafer A, et al. An ASO therapy for Angelman syndrome that targets an evolutionarily conserved region at the start of the UBE3A-AS transcript. Sci Transl Med. 2023;15:eabf4077.
Google Scholar
-
Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, et al. Comparison of Cas9 activators in multiple species. Nat Methods. 2016;13:563–7.
Google Scholar
-
O’Geen H, Ren C, Nicolet CM, Perez AA, Halmai J, Le VM, et al. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017;45:9901–16.
Google Scholar
-
Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015;12:401–3.
Google Scholar
-
Guhathakurta S, Kim J, Adams L, Basu S, Song MK, Adler E, et al. Targeted attenuation of elevated histone marks at SNCA alleviates alpha-synuclein in Parkinson’s disease. EMBO Mol Med. 2021;13:e12188.
Google Scholar
-
Wilson C, Chen PJ, Miao Z, Liu DR. Programmable m(6)A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol. 2020;38:1431–40.
Google Scholar
-
Liu XM, Zhou J, Mao Y, Ji Q, Qian SB. Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol. 2019;15:865–71.
Google Scholar
-
Tsai TF, Jiang YH, Bressler J, Armstrong D, Beaudet AL. Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Hum Mol Genet. 1999;8:1357–64.
Google Scholar
-
Resnick JL, Nicholls RD, Wevrick R, Prader-Willi Syndrome Animal Models Working G. Recommendations for the investigation of animal models of Prader-Willi syndrome. Mamm Genome. 2013;24:165–78.
Google Scholar
-
Kummerfeld DM, Raabe CA, Brosius J, Mo D, Skryabin BV, Rozhdestvensky TS. A comprehensive review of genetically engineered mouse models for Prader-Willi syndrome research. Int J Mol Sci. 2021;22:7.
Google Scholar
-
Relkovic D, Doe CM, Humby T, Johnstone KA, Resnick JL, Holland AJ, et al. Behavioural and cognitive abnormalities in an imprinting centre deletion mouse model for Prader-Willi syndrome. Eur J Neurosci. 2010;31:156–64.
Google Scholar
-
Dubose AJ, Smith EY, Yang TP, Johnstone KA, Resnick JL. A new deletion refines the boundaries of the murine Prader-Willi syndrome imprinting center. Hum Mol Genet. 2011;20:3461–6.
Google Scholar
-
Yang T, Adamson TE, Resnick JL, Leff S, Wevrick R, Francke U, et al. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat Genet. 1998;19:25–31.
Google Scholar
-
Jiang YH, Pan Y, Zhu L, Landa L, Yoo J, Spencer C, et al. Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3. PLoS One. 2010;5:e12278.
Google Scholar
-
Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811.
Google Scholar
-
Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, et al. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest. 2015;125:2069–76.
Google Scholar
-
Wang T, van Woerden GM, Elgersma Y, Borst JGG. Enhanced transmission at the calyx of held synapse in a mouse model for Angelman syndrome. Front Cell Neurosci. 2017;11:418.
Google Scholar
Acknowledgements
We would like to thank Emily Qian for her comments and editing for the manuscript. The research in Yong-hui Jiang’s lab is supported by National Institute of Health (MH117289 and HD088007), Foundation for Angelman Syndrome Therapeutics, and National Research Foundation of Korea (NRF-2020R1A6A3A03037861).
Author information
Authors and Affiliations
Contributions
SEW and YHJ wrote text and designed the figures for the manuscript. All authors reviewed and edited the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
YHJ is a cofounder for a new startup company of Couragene. However, Couragene did not have any direct role in this review. The other author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Wang, S.E., Jiang, Yh. Novel epigenetic molecular therapies for imprinting disorders.
Mol Psychiatry (2023). https://doi.org/10.1038/s41380-023-02208-7
-
Received: 12 February 2023
-
Revised: 21 July 2023
-
Accepted: 27 July 2023
-
Published: 25 August 2023
-
DOI: https://doi.org/10.1038/s41380-023-02208-7