
Congenital disorders
First report of bovine viral diarrhea virus subgenotypes 1d and 1e in southern Chile
Sep
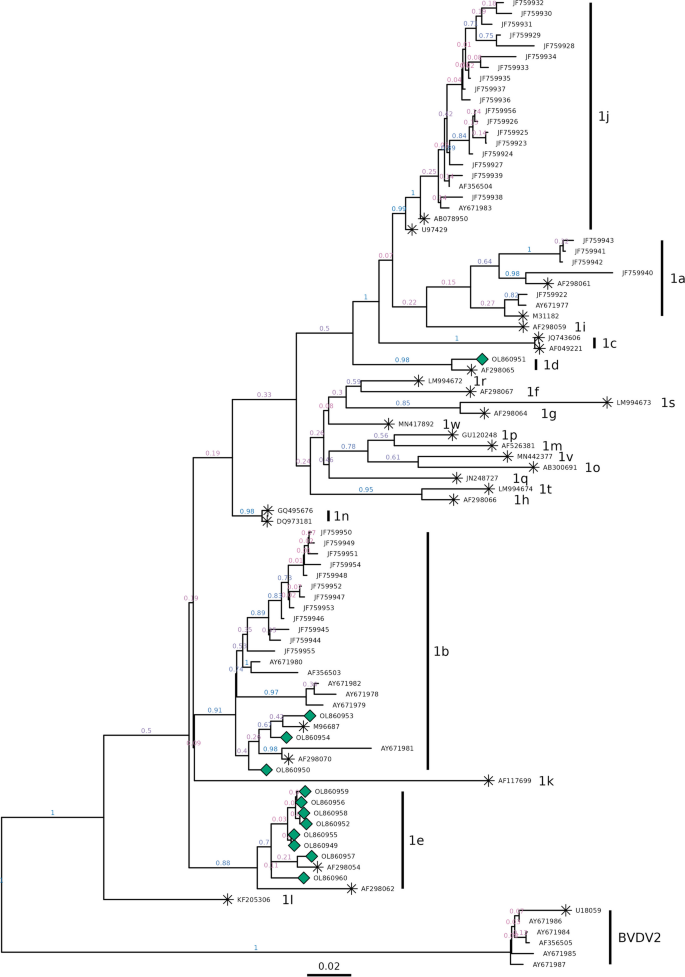
From the 24 antigenically positive serum samples, 12 amplicons were obtained for sequencing procedures. The low recovery of amplicons is probably due to RNA degradation consecutive to the delay between the first analysis of the samples and the reception of them in our laboratories, what led to repeated frozen-thaw cycles [28]. According to the BLAST analysis of the obtained sequences, all the samples belonged to Pestivirus bovis, from which eight corresponded to subgenotype 1e, three samples were classified as subgenotype 1b and one as 1d (Table 1). The phylogenetic tree confirmed the species classification of the samples, as well as their subgenotypes (Fig. 1). From the resulting consensus tree, eight samples were classified as clade 1e by 0.70 of posterior probability (pp) and forming a separate cluster from the strains earlier reported in Chile (Fig. 1, pp = 0.89). The distance with the closest clade, which corresponds to sequences of subgenotypes 1b (pp = 0.92), showed 0.062 changes per nucleotide.
Bayesian consensus phylogenetic tree based on the 5’UTR sequences of Chilean isolates (ref [6] and [21], without symbol), references strains (identified with *) and the 12 strains under study from the Aysén region (green diamond). Values behind the nodes indicate posterior probability values, with a colour gradient between low (0.0) in red to high (1.0) in blue. Subgenotypes are indicated in the right side
Three of our samples from Aysén (i.e., OL860953, OL860954 and OL860950) belonged to clade 1b. These samples cluster to a subclade together with M96687 from Osloss (Germany), AF298070 from Austria and AY671981 from Central Chile (pp = 0.92). The BLAST analysis revealed the highest similarity with strains reported from Argentina, United Kingdom and Italy (Additional file 2: Table S1). The strain from Argentina was recently registered and corresponds to a sample taken in 2019, posterior to our sampling date. These results seem to indicate that 1b strains may have a European origin, but it is difficult to explain the route of transmission, as importation of live animals is not a common practice for Chile, and semen used for artificial insemination comes mainly from U.S.A. and Canada and must be certified free of BVDV and other pathogens. Only one of analyzed samples (i.e., OL860951) belongs to the clade 1d and has over 97% identity with strains from Brazil and China (Additional file 2: Table S1). The Brazilian strain corresponds to a recent sampling while the Chinese strain was collected ten years ago.
The existence of discrete genetic units based on mPTP analysis confirms the units 1a, 1b, 1d, 1e, 1j and Pestivirus tauri. Strains belonging to subgenotypes 1f, 1g, 1h, 1m, 1o, 1p, 1q, 1r, 1s, 1t, 1v and 1w form a single unit (Additional file 1: Fig. S1). Genetic diversity analyzed using a haplotype network based on haplogroups highlights the presence of one haplotype for 1e unit, gathering our 8 samples from Aysén together with AF298054, and well-defined units based on differentiated haplotypes proximity (Fig. 2). SDT pair-wise ranking of the sequences showed an identity between 99.1 and 100% for samples 1e; 94.8 to 98.8% for samples 1b; while the sample 1d had a mean identity of 87.5% and 87.4% with samples 1e and 1b, respectively. As for the comparison between samples 1e and 1b, the identity ranged from 89.0 to 90.5%.

Median-joining haplotype network tree based on the 5’UTR sequences of Chilean isolates (ref [6] and [21], without symbol), references strains (identified with*) and of the 12 strains under study from the Aysén region (green diamond). Haplotypes are identified by the numbers in the circles; circle’s size is proportional to its frequency. Hatches on the lines represent mutational steps. Colors within the circles represent the strains where haplotypes were found
Genotyping the 5’UTR is commonly used as it is a highly conserved region for BVDV [1, 34]. The sequencing of the isolates from the Aysén region allowed reporting BVDV subgenotypes 1e and 1d for the first time in Chile. Previously, based on 5’UTR sequencing, subgenotypes 1a, 1b, 1c and Pestivirus tauri had been reported in sampled animals from the central regions of Chile between 1993 and 2001[24]. Later, in samples taken between years 2003 and 2007 from central and south-central regions of the country, the presence of subgenotypes 1a and 1b were confirmed, adding subgenotype 1j, based on the sequencing of 5’UTR and E2 genomic sequences [7]. Reports from Argentina only showed the presence of subgenotypes 1a, 1b and Pestivirus tauri [14, 22], even though recently, subgenotypes 1e and 1i were reported but with only n = 1 for each strain [31]. The major risk of contact with animals from this neighbor country may occur because of the use of summer fields located along the border. On the continent, only in Brazil were reported strains from subgenotypes 1d and 1e previously [29, 35], and there are not movements of cattle registered between Brazil and Chile, though, it is unlikely that a transmission occurs through this way. But recently, a new 1e strain from Brazil was registered in GenBank data base and it has between 99.56 and 100% identity with the 1e strains from Aysén (Additional file 2: Table S1). The 1e strains under study also have high identity (between 99.17 and 100%) with strains from United Kingdom, Switzerland, and Italy (Supplementary Table S2), which indicates again the possibility of a European origin. Besides, in our evaluation, subgenotype 1b was found, in agreement with previous reports in the country [7, 24]. This result was expected, since subgenotype 1b is the most represented subgenotype worldwide [35], even though, in this case, a higher frequency (66.7%) was observed for subgenotype 1e versus 25% for 1b.
In Chile, even if there are some producers who vaccinate their cattle against BVDV, the percentage of effectively vaccinated animals is unknown. The only commercial vaccine available in the country is inactivated and contain strains 1a and 2 (Cattle Master Gold FP5®) and until two years ago, also another vaccine containing strains 1a and 1b (Cattle Master®). In a study carried out in the USA, it was suggested that the regression over time of one subgenotype in a particular region might be related to the use of vaccine strains targeting the same subgenotype, which would induce a change in dominance of the current subgenotypes [26]. This fact could partly explain the lower frequency of subgenotype 1b in the samples studied, although a larger number of samples as well as comparison with samples from the region in previous years is required to strongly corroborate this explanation. Moreover, studies have shown that different vaccines, including killed and live attenuated vaccines, composed with strains 1a or 1b, have varying levels of neutralizing antibodies against heterologous subgenotypes of 1a or 1b strains [9, 30]. Additionally, these vaccines have been found to have a total absence of neutralizing antibodies against 1e strains [30]. This highlights the importance of incorporating circulating strains in the formulation of new BVDV vaccines to promote multivalent protection.
Phylogenetic trees inferred from single gene sequences are gene trees rather than species trees, although hierarchical relationships are expected to be consistent with species trees [36]. In this case, the phylogenetic tree obtained represents the intraspecific variations of the sequences analyzed. Network analysis is a tool used to establish and visualize the number of sites that are different between two sequences. For closely related sequences, this represents a good estimation of the number of mutations separating both sequences from their common ancestors [17]. Here, the diversity of strains found, and the high number of mutations observed are indicators of the existence of a pathway of virus introduction related with another genetic origin that was not present in the region of Aysén. Due to the low number of samples analyzed here, it is necessary to integrate more data, like the sequences of multiple genes, to validate the relationships [36].
The present results, added to previous studies, reveal a great diversity of BVDV subgenotypes circulating in Chile. In addition, it provides preliminary evidence of the variability of strains over time and how the continuous application of strain-specific vaccines can modulate the emergence of new strains. Although sampling was limited, this is the first report of subgenotypes 1d and 1e in Chile, both very well supported by Bayesian phylogenetic tree and mPTP analyses. The distance found between these strains and others reported in Chile evidences the continuous need to expand the monitoring in herds and to deepen the analysis of sequences spatially and retrospectively, to define the entry and transmission pathway of the virus, as well as its behavior in terms of mutation and dominance. These results, together with another random sampling, should be considered in the design of new vaccines that will contribute to the successful implementation of viral control and eradication programs in the area.