
Cancer and neoplasms
Polycythemia vera & newly diagnosed multiple myeloma
Sep
Introduction
Polycythemia vera (PV) is the most common Philadelphia chromosome-negative myeloproliferative neoplasms, characterized by a hypercellular bone marrow status that results in increased numbers of erythroid, megakaryocytic and granulocytic precursor cells and an absolute increase in hematocrit.1–3 Most patients (>95%) with PV presented acquired Janus kinase 2 (JAK2) gene mutation. Major complications include arterial and venous thrombosis, hemorrhage, and the risk of leukemic transformation.1–4 Multiple myeloma (MM) is a neoplastic plasma-cell disorder characterized by plasma cell clonal proliferation in the bone marrow microenvironment, monoclonal protein formation, and associated organ dysfunction.5,6
The report on the simultaneous coexistence of PV and MM is rare, with about twenty-four cases reported in the literature.7–15 Most of these reports included patients who had antecedent or concomitant PV or who already developed secondary myeloma after radiation therapy of PV for years. We describe a female patient who presented with NDMM and subsequently met diagnostic criteria of PV well established by extensive laboratory workup for the first time.3 Additionally, we reviewed previous studies of concurrent PV with NDMM to investigate the clinical features and possible interactions between the two hematopoietic disorders.
Materials and Methods
A case report of concurrent PV with NDMM in a Chinese female patient is presented. Hematological evaluations included bone marrow (BM) aspiration smear, flow cytometry (FCM), myeloproliferative neoplasms specific fusion gene testing, fluorescence in situ hybridization (FISH), cytogenetic karyotype analysis, biopsy slice, immunohistochemistry and reticular fiber staining. Systemic evaluations included laboratory examinations for blood routine test, biochemistry, coagulation, erythrocyte sedimentation rate (ESR), immunoglobulin levels, urinalysis and 24-hour urinary protein quantity, immunofixation and electrophoresis of serum and urine, serum erythropoietin (EPO) level. X-ray of skull, pelvis and limb bones. In addition, the magnetic resonance imaging (MRI) of vertebra was also performed.
We completed a PubMed database search of articles in English up to January 2020. The search was conducted using keywords as following: “polycythemia vera”, “multiple myeloma”, “concurrence” and “simultaneous”. The specific diagnostic and response criteria were previously defined.2,5,6 We evaluated all the literature and identified reported cases of PV concurrence with MM to investigate the clinical features and discuss the clinical features and possible pathogenetic interactions.
Results
A 70-year-old woman was admitted to Department of Hematology with a 2-month history of lower backache. She had a 5-year history of cerebral infarction without sequelae, but never had oral anticoagulation or antiplatelet agents. There was no family history of erythrocytosis or any myeloproliferative disease. She had no trauma history. Physical examination revealed a plethoric face, lumbar percussion pain, impalpable liver, spleen and otherwise unremarkable findings. White blood cell count (WBC) was 19.4×109/L (normal range 3.5~9.5), with normal differential WBC, platelets (PLT) 425×109/L (normal range 125~350), hematocrit 54.1% (normal range 35%~45%) with hemoglobin (HGB) 16.5g/dL (normal range 11.5~15). The patient’s serology was negative for viral hepatitis. The initial laboratory findings showed an albumin (ALB) of 3750mg/dL (normal range 4000~5500), globulin 4140mg/dL (normal range 2000~4000), uric acid (UA) 6.5mg/dL (normal range 2.6~6.0), β2-micro globulin (β2-MG) 4.44mg/L (normal range 0.97~2.64), calcium 8.76mg/dL (normal range 8.44~10.08) and lactic dehydrogenase (LDH) 394U/L (normal range 120~250). The ESR was 9mm/h (normal range <20). The prothrombin time (PT) was 11.8 seconds (normal range 9.4~12.5) and the prothrombin activity (PTA) was 89% (normal range 80%~120%). The international normalized ratio (INR) was 1.07 (normal range 0.8~1.2), fibrinogen was 3.12g/L (normal range 2.38~4.98), and activated partial thromboplastin time (APTT) was 32 seconds (normal range 25.4~38.4). The immunoglobulin test showed that IgG 1.18g/dL (normal range 0.75~1.5), IgA 2.19g/dL (normal range 0.1~0.5), IgM 0.085g/dL (normal range 0.046~0.3). Urinalysis and 24-hour urinary protein quantity revealed negative. Serum immunofixation and electrophoresis showed an IgA-κ monoclonal protein with quantification of 1.26 g/dL. Urine immunofixation and electrophoresis showed Bence-Jones protein negative. Serum-free κ light chains 17.8mg/L (normal range 3.3~19.4), free λ light chains 20.6mg/L (normal range 5.71~26.3), κ/λ light chain ratio 0.86 (normal range 0.26~1.65). Urine free κ light chains 59.7mg/L (normal range 0.39~15.1), free λ light chains 5.73mg/L (normal range 0.81~10.1), κ/λ light chain ratio 10.42 (normal range 0.461~4.0). Serum EPO level was 4.08mIU/mL (normal range 2.59~18.5).
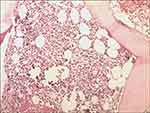
The BM cell morphology indicated hypercellularity, while the immature plasma cells percentage was 3%. The BM core biopsy (Figure 1) revealed 70% cellularity with 10% κ light chain restricted plasma cells. Reticular fiber staining was negative. Immunohistochemistry staining revealed as following: CD138(+), CD38(+), CD56(+), CD19(-), CD117(-), CD20(-), κ(+), λ(-). BM sample was submitted for FCM testing, which revealed an abnormal plasma-cell population (0.1%) strongly expressing CD38, CD138, CD56, CD81 and cytoplasmic κ, partially expressing CD27 (58.33%) and CD117 (87.68%), with no expression of cytoplasmic λ, CD19, CD20. Marrow cytogenetics showed 46, XY, but FISH (plasma cells enrichment by CD38+ magnetic beads) analysis showed 10.5% of nuclei with 1q21 amplification, 9.5% of nuclei with 13q14 (RB1) deletion and 8.0% of nuclei with 13q14.3 (D13S319) amplification. The allele-specific PCR of BM sample revealed that JAK2 V617 mutation positive, while BCR-ABL1, MPL and CALR mutation negative. The X-ray of skull, pelvis and limb bones revealed negative destruction or lytic lesions. The MRI of vertebra revealed compression fracture of the second lumbar vertebrae. In conclusion, the patient was diagnosed with IgA-κ MM, Durie-Salmon stage IIIA, revised international scoring system II.
|
Figure 1 Low power BM core biopsy (×20) demonstrating hypercellularity consistent with a myeloproliferative disorder, and revealed 70% cellularity with 10% κ light chain restricted plasma cells. |
The patient subsequently received a total of five cycles regimen of bortezomib, cyclophosphamide and dexamethasone. Meanwhile, oral antiplatelet agent aspirin was also given for prophylaxis of thrombosis. The initial response evaluation achieved partial response, while erythrocytosis was well-controlled until loss of follow-up (14-JAN-2020).
Discussion
PV and MM are both clonal disorders of the hematopoietic stem cell. Nevertheless, the coexistence of PV and MM occurring simultaneously in the same patient is uncommon. Most of the previous reports indicated that MM was secondary of PV, particularly after long-term treatment with radiation.7–14,16 Concurrent PV with NDMM is extremely rare. Lawrence and Rosenthal first described a case of concurrent diagnosis of MM and PV in 1949.7 Most of cases reported were before 2005, when genetic mutation detection or immunofixation electrophoresis were not available. Our present case is the first to adopt genetic testing confirming JAK2 V617F mutation positive and determination confirming the decrease of serum EPO level in a PV patient with NDMM.
With a review of previous medical literature, we identified eight cases of these two concurrent hematological neoplasms. All cases were of uncommon simultaneous occurrence.7,11–14,16 The details of eight similar cases (including our present case) are summarized in Table 1. Among eight patients, the median age was 71 years (range, 41~80 years) with no gender predominance (four females and four males). These cases were reported before 1990s or earlier, with limited serum electrophoresis data. Only four cases had immunofixation and electrophoresis results, which showed the monoclonal protein IgG (2 cases) and IgA (2 cases). Two cases were simultaneous occurrent PV with smoldering myeloma: the management were venesection or phlebotomy alone accordingly. Eight cases had at least one systemic manifestation, including dizziness, fatigue, weakness, light headedness, plethoric face or shortness of breath, which are common in PV and MM. Four cases revealed compression fracture of lumbar vertebrae or lytic lesions; five cases exhibited splenomegaly or hepatomegaly. In addition, there were three cases with history of thrombosis, indicating that PV may develop ahead of MM. Treatment regimens were unavailable in two cases’ profiles, while other cases were treated with combination therapy with melphalan, procarbazine, cyclophosphamide and prednisone. Only our patient underwent the chemotherapy including proteasome inhibitor. Remarkably, concomitant polycythemia resolved after initiation of therapy for myeloma.
 |
Table 1 Patients with Concurrence of PV and MM: Review of Literature |
Anemia is one of the major clinical manifestations of MM and can be observed in 73% of MM patients.13 Our patient revealed uncommon initial erythremia, hence the allele-specific PCR of BM samples confirmed JAK2 V617 mutation positive. It demonstrated that abnormal erythremia, hepatosplenomegaly and thrombosis history suggested comorbidity of PV with NDMM. Furthermore, concomitant erythremia could be resolved after bortezomib-based chemotherapy for MM.
Previous investigators have postulated the mechanism of the concurrence of myeloma and polycythemia, including coincidence, clonal evolution, and therapy-induction.14–16 Both PV and MM are clonal diseases of hematopoietic stem cell, which represent clonal origin at a much earlier stage in differentiation involving abnormal proliferation of both myeloid and lymphoid cell lines. It is known that JAK2 mutation increases the risk of developing malignancies including lymphoma and solid cancers,17–19 which might explain the development of NDMM in our case. We also realize that most of PV patients had a history of radiation therapy, which was speculated to be the pathogenesis for development of myeloma. However, the pathogenesis needs to be established in how PV evolved into MM in further investigations. To the best of our knowledge, our study is the first patient with positive JAK2 V617 mutation and decreased EPO level in such concurrence, while previous reports failed to provide genetic mutation profiles for the diagnosis of PV.17
Due to lack of such concurrence of PV and MM cases, clinical treatment is relatively unestablished. In the current study, our patient received a bortezomib-based chemotherapy regimen, which seemed to be effective on PV. In addition, JAK2 V617F mutation is very infrequent in human MM despite the fact that constitutional activation of JAK2 kinase as well as STAT3 phosphorylation is frequently seen in MM patients.20 It suggested that the JAK2 inhibitor as Ruxolitinib may be a potential approach, especially for patients with splenomegaly.21
In conclusion, concurrent PV with NDMM may coexist simultaneously in the same patient, though it is extremely rare. The present case is the first patient of NDMM with diagnosis of PV confirmed by positive JAK2 V617F mutation. Abnormal erythremia, hepatosplenomegaly and thrombosis history suggested comorbidity of PV with NDMM. Concomitant polycythemia can be resolved after initiation of chemotherapy for myeloma. Whereas the pathogenesis of these two entities remains to be further investigated.
Data Sharing Statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for Publication
Written informed consent for publication of identifying images or other personal or clinical details was obtained from the patient prior to study commencement. The institutional approval was not required to publish the case details.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Spivak JL. Polycythemia vera. Blood. 2019;133(25):2630–2631. doi:10.1182/blood-2019-04-901132
2. McMullin MF, Wilkins BS, Harrison CN. Management of polycythaemia vera: a critical review of current data. Br J Haematol. 2016;172(3):337–349. doi:10.1111/bjh.13812
3. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–770. doi:10.1200/JCO.2010.31.8436
4. Spivak JL. How I treat polycythemia vera. Blood. 2019;134(4):341–352. doi:10.1182/blood.2018834044
5. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548. doi:10.1016/S1470-2045(14)70442-5
6. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197–2208. doi:10.1016/S0140-6736(14)60493-1
7. Lawrence JH, Rosenthal RL. Multiple myeloma associated with polycythemia; report of four cases. Am J Med Sci. 1949;218(2):149–154. doi:10.1097/00000441-194908000-00005
8. Inase N, Shichiri M, Marumo F. Secondary polycythemia associated with multiple myeloma. Jpn J Med. 1989;28(3):396–398. doi:10.2169/internalmedicine1962.28.396
9. Chang H, Shih LY. Concurrence of multiple myeloma and idiopathic erythrocytosis. Acta Clin Belg. 2009;64(5):434–435. doi:10.1179/acb.2009.071
10. Hutchison EJ, Taverna JA, Yu Q, Yeager AM. Polycythaemia: an unusual presentation of multiple myeloma. BMJ Case Rep. 2016;bcr2016216686. doi:10.1136/bcr-2016-216686
11. Kelsey PR, Patel K. Coexistence of polycythaemia vera with indolent myeloma in the same patient. Hematology. 1997;2(2):139–142. doi:10.1080/10245332.1997.11746329
12. Fink L, Bauer F, Perry JJ. Coincidental polycythemia vera and multiple myeloma: case report and review. Am J Hematol. 1993;44(3):196–200. doi:10.1002/ajh.2830440311
13. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21–33. doi:10.4065/78.1.21
14. Maeda K, Abraham J. Polycythemia associated with myeloma. Am J Clin Pathol. 1984;82(4):501–505. doi:10.1093/ajcp/82.4.501
15. Heinle EW, Sarasti HO, Garcia D, Kenny JJ, Westerman MP. Polycythemia vera associated with lymphomatous diseases and myeloma. Arch Intern Med. 1966;118(4):351–355. doi:10.1001/archinte.1966.00290160051010
16. Franzén S, Johansson B, Kaigas M. Primary polycythaemia associated with multiple myeloma. Acta Med Scand Suppl. 1966;445:336–343. doi:10.1111/j.0954-6820.1966.tb02380.x
17. Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8(2):15. doi:10.1038/s41408-018-0054-y
18. Vannucchi AM, Masala G, Antonioli E, et al. Increased risk of lymphoid neoplasms in patients with Philadelphia chromosome-negative myeloproliferative neoplasms. Cancer Epidemiol Biomarkers Prev. 2009;18(7):2068–2073. doi:10.1158/1055-9965.EPI-09-0353
19. Nielsen C, Birgens HS, Nordestgaard BG, Kjaer L, Bojesen SE. The JAK2 V617F somatic mutation, mortality and cancer risk in the general population. Hamatologica. 2011;96(3):450–453. doi:10.3324/haematol.2010.033191
20. Huang Q, Li X, Chen W, Weiss LM. Absence of JAK-2V617F point mutations in multiple myeloma. Leukemia. 2007;21(4):813–814. doi:10.1038/sj.leu.2404551
21. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. doi:10.1056/NEJMoa1409002