
Congenital disorders
Allelic hierarchy for USH2A influences auditory and visual phenotypes in South Korean patients
Nov
Abstract
When medical genetic syndromes are influenced by allelic hierarchies, mutant alleles have distinct effects on clinical phenotypes. Genotype–phenotype correlations for Usher syndrome type 2 (USH2) suggest that the USH2A gene exhibits an allelic hierarchy. Here, we analyzed the phenotypes and genotypes of 16 South Korean patients with USH2A biallelic variants to investigate an allelic hierarchy from audiological and ophthalmological perspectives. Using whole exome and genome sequencing, 18 mutant alleles, including 4 novel alleles, were identified and implicated in USH2A-related disorders. Truncated alleles were linked to earlier onset of subjective hearing loss and more severe thresholds; biallelic truncated alleles had more severe effects. Truncated alleles were also associated with retinal structure degeneration and severe functional deterioration. However, younger patients (aged < 16 years) did not exhibit overt retinitis pigmentosa even when they had biallelic truncated alleles, suggesting that USH2A-related USH2 can mimic nonsyndromic hearing loss. For truncated alleles, there was a clear correlation between mean hearing threshold and 30-Hz flicker electroretinography implicit time. This study provides the first evidence of an USH2A-related allelic hierarchy among South Korean patients; our data yield valuable insights concerning the natural courses of clinical phenotypes and how genotype-based therapies may be used.
Introduction
Usher syndrome, the leading cause of deaf-blindness, is clinically and genetically heterogeneous; it is classified into three primary subtypes based on the severity and onset of subjective hearing loss, along with the presence of vestibular dysfunction1. The genes responsible for Usher syndrome encode proteins of the retina and inner ear with key roles in photoreceptor maintenance, as well as sensory hair cell development and function2,3. These genes form the Usher interactome, which regulates clinical phenotypes via functional epistasis4,5. Among the disease subtypes, Usher syndrome type 2 (USH2) is the most prevalent, constituting more than half of all cases6. USH2 gene loci include USH2A (usherin), USH2C (ADGRV1), and USH2D (whirlin)7. Of these, USH2A (OMIM 608400), located on chromosome 1q41, is implicated in up to 85% of USH2 cases8. Biallelic variants of USH2A have been linked to clinical heterogeneity in USH2A-related disorders, including USH2, nonsyndromic retinitis pigmentosa (RP), and nonsyndromic sensorineural hearing loss (SNHL)9.
Audiological manifestations of USH2 typically precede visual symptoms and signs7. Congenital hearing loss is detected via screening of newborn infants, which enables clinicians to identify syndromic deafness mimics (e.g., Usher syndrome) prior to the emergence of overt syndromic phenotypes10. The audiological profiles range in severity from moderate to severe, with a linear decrease that is particularly noticeable at high frequencies11,12. Additionally, USH2 causes RP with clinical symptoms that include nyctalopia and visual field constriction13. As the disease progresses, central vision is gradually lost, leading to blindness. Clinical signs through image evaluation include the collapse of retinal layers found in optical coherence tomography (OCT), optic nerve pallor, the development of bony spicules found in fundus photo, and the appearance of hyper-autofluorescent rings in the paramacular region13,14. Alterations in electroretinogram (ERG) and visual acuity are also accompanied. Because RP is generally progressive, age is a major risk factor. Although retinal degeneration can begin in childhood, disease progression may be slow and retinal phenotypes may not be noticed15,16; the median age at diagnosis of non-syndromic RP is ~ 35 years, when patients become aware of the problem and seek medical advice15. In case of syndromic RP, which USH2A-related consists most of the patients, the median age at diagnosis is earlier at 18–30 years17. However, in both types, it is still difficult for patients to notice retinal phenotypes in their childhood or adolescence.
The integration of phenotypic and genotypic information has improved the broader understanding of USH2A-related disorders, particularly concerning prediction of the clinical course of disease and selection of targeted therapies. The results of recent studies suggest that the USH2A gene exhibits an allelic hierarchy; additionally, different mutant alleles or their combinations have distinct effects on clinical phenotypes18. In particular, at least one nonsyndromic RP-specific allele and other missense variants with few deleterious effects are associated with isolated RP or RP with progressive hearing loss19, which is present in 86% of patients with USH2A variant-related nonsyndromic RP9. Moreover, at least one particular truncated variant of USH2A causes severe retinopathy20; when truncated variants of USH2A are combined, the resulting hearing loss may require cochlear implantation (CI)12,21.
Despite advances in the understanding of genotype–phenotype correlations for USH2, no studies have investigated the allelic hierarchy of USH2A-related disorders from audiological and ophthalmological perspectives in South Korean patients. Differences in genotypes among ethnic groups may alter the impact of mutant allele combinations, and previous data regarding allelic hierarchies may not be directly applicable to all patients with USH2A-related disorders22. Here, we investigate the links between genotypes and clinical phenotypes in South Korean patients, including audiological and ophthalmological function and retinal structure. Additionally, we evaluate the effects of truncated alleles from audiological and ophthalmological perspectives, suggesting an allelic hierarchy.
Results
Genotypes of USH2A-related disorders
We identified 14 biallelic variants in USH2A, either homozygous or compound heterozygous, in a trans configuration. The segregation of these variants was confirmed by Sanger sequencing. Overall, 18 mutant alleles were implicated in the diagnosis of USH2A-related phenotypes, including c.251G > A:p.Cys84Tyr, c.2209C > T:p.Arg737*, c.2802 T > G:p.Cys934Trp, c.4372C > T:p.Arg1578Cys, c.4858C > T:p.Gln1620*, c.7120 + 1475A > G, c.8232G > C:p.Trp2744Cys, c.85559-2A > G, c.10593del:p.Ile3532Phefs*18, c.10712C > T:p.Thr3571Met, c.10724G > T:p.Cys3575Phe, c.11156G > A:p.Arg3719His, c.12708 T > A:p.Cys4236*, c.13112_13115del:p.Gln4371Argfs*19, c.14134-3169A > G, c.13964 T > C:p.Leu4655Pro, c.14835del:p.Val4946Trpfs*4, and c.14911C > T:p.Arg4971* (Table 1 and Supplementary Fig. S1). The amino acid residues were highly conserved in orthologs from several species (Supplementary Fig. S1), a finding consistent with the corresponding high Genomic Evolutionary Rate Profiling (GERP + +) score. Two variants, c.2802 T > G:p.Cys934Trp and c.8559-2A > G variants, were present more than twice. Consistent with our findings, these two variants have been recurrently identified and are recognized as East Asian-specific founder and mutational hotspot alleles associated with USH2A gene23. Four variants were novel, including two missense variants, one frameshift variant, and one deep intronic variant. The frameshift variant p.Ile3532Phefs*18 had a premature termination codon, which resulted in nonsense-mediated mRNA decay. This variant was extremely rare in population databases, including the Korean Reference Genome Database, which is based on genetic data from South Koreans. Furthermore, co-segregation analyses confirmed the presence of this variant in trans with a known pathogenic variant (c.8559-2A > G). Based on the American College of Medical Genetics and the Association for Molecular Pathology (ACMG/AMP) guidelines24,25, this novel frameshift variant was classified as pathogenic (PVS1, PM2, PM3). The novel missense variants p.Cys3575Phe and p.Leu4655Pro, located in the fibronectin type III-20 and fibronectin type III-32 domains, respectively, were also extremely rare in population databases and occurred in trans with a pathogenic variant. In silico analysis, using Combined Annotation Dependent Depletion26, predicted that these variants would be pathogenic. Based on the ACMG/AMP guidelines24,25, the novel missense variants p.Cys3575Phe and p.Leu4655Pro were classified as likely pathogenic (PM2, PM3, PM5, PP4) and variants of uncertain significance (PM2, PM3, PP4), respectively. Furthermore, two deep intronic variants, c.14134-3169A > G and c.7120 + 1475A > G, were identified by trio-based whole-genome sequencing (WGS) and bioinformatics analyses. The previously reported c.14134-3169A > G variant in intron 64 led to pseudo-exon activation, which resulted in a truncated protein27. The novel deep intronic variant c.7120 + 1475A > G in intron 37 was predicted to introduce pseudo-exon activation. This newly inserted pseudo-exon had a premature termination codon, which probably resulted in a truncated translation product. We have reclassified the conflicting or variants of uncertain significance documented in the ClinVar database, using the ACMG/AMP guidelines24,25. Upon evaluation using either the ClinVar database or the ACMG/AMP guidelines, 16 of the 18 variants (with the exceptions of c.251G > A:p.Cys84Tyr and c.13964 T > C:p.Leu4655Pro) were determined as either pathogenic or likely pathogenic (Table 1). The genotype information of the disease-causing USH2A variants, mapped to both hg19 and hg38, is described in Supplementary Table 1.
To investigate the allelic hierarchy of USH2A-related disorders, we categorized patients with USH2A biallelic variants into three groups based on their truncated and nontruncated mutant alleles: group 1 consisted of individuals with two truncated mutant alleles; group 2 consisted of individuals with one truncated and one nontruncated mutant allele; and group 3 consisted of individuals with two nontruncated mutant alleles (Supplementary Table 2).
Audiological phenotypes and allelic hierarchy
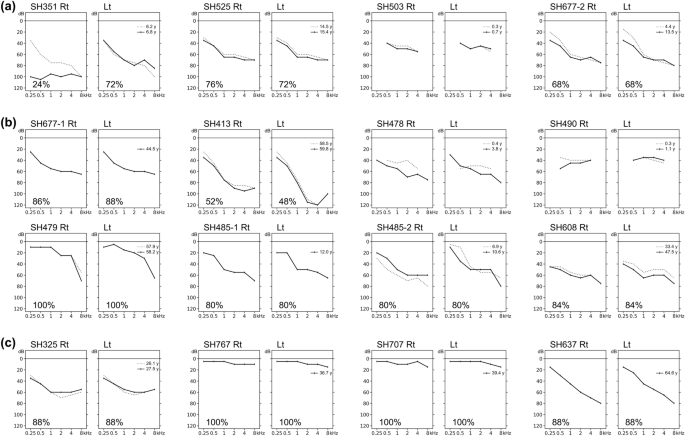
The audiological characteristics of our cohort are shown in Table 2; the corresponding audiograms are shown in Fig. 1. Based on the latest audiograms, three patients (18.8%) had normal hearing (SH479, SH767, and SH707), indicating nonsyndromic RP. The remaining 13 patients (81.2%) had varying degrees of SNHL; this was typically mild-to-moderate at low frequencies and severe-to-profound at high frequencies. Among these 13 patients, six had moderate SNHL (46.2%), five had moderately severe SNHL (38.5%), and two had severe SNHL (15.4%). Subjective onset of hearing loss was congenital in six (46.2%), prelingual in two (15.4%), and postlingual in five (38.5%) patients. All but one patient (SH351) had symmetric bilateral hearing loss that deteriorated at higher frequencies (10 of 12, 83.3%), or they exhibited a horizontal trace pattern (2 of 12, 16.7%). Consistent with the findings in previous studies21, during the follow-up period, which ranged from 0.3 to 14.1 years with a median duration of 1.3 years, all 11 patients who underwent hearing evaluations had milder progressive hearing loss. One patient (SH351) with biallelic truncated variants underwent unilateral CI in their worse ear before the age of 13 years. Neither congenital cytomegalovirus infection nor inner ear anomalies underlying asymmetric hearing loss were identified in this patient (SH351). Most patients with confirmed hearing loss used hearing aids.
Audiograms of 16 patients with biallelic Usher syndrome type 2A (USH2A) variants. (a) Four patients in group 1. (b) Eight patients in group 2. (c) Four patients in group 3. Speech discrimination scores are indicated in lower left corners, except when the test was not performed because the patient was too young.
The mean hearing thresholds were 59.7 dB, 51.1 dB, and 29.1 dB in group 1 (two truncated alleles), group 2 (one truncated and one nontruncated allele), and group 3 (two nontruncated alleles), respectively (Fig. 2a). The thresholds were consistently highest across frequencies in group 1 and lowest in group 3. We recorded mean hearing thresholds for group 1 versus group 2 versus group 3 at 250 Hz (35.0 vs. 24.3 vs. 15.0, p = 0.015 by Kruskal–Wallis rank sum test), 500 Hz (46.3 vs. 36.9 vs. 20.0, p = 0.021 by Kruskal–Wallis rank sum test), 1000 Hz (62.5 vs. 50.6 vs. 27.5, p = 0.005 by Kruskal–Wallis rank sum test), 2000 Hz (65.0 vs. 56.9 vs. 32.5, p = 0.024 by Kruskal–Wallis rank sum test), 4000 Hz (65.0 vs. 60.0 vs. 36.3, p = 0.032 by Kruskal–Wallis rank sum test), and 8000 Hz (78.3 vs. 75.7 vs. 41.3, p = 0.013 by Kruskal–Wallis rank sum test). Patients with at least one truncated allele (groups 1 and 2) tended to have more severe hearing loss than patients with biallelic nontruncated alleles (group 3; Fig. 2b); patients with one truncated allele had higher hearing thresholds than patients with biallelic nontruncated alleles (Fig. 2c). Nonsyndromic RP was more common among patients in group 3 than among patients in the other groups. All patients in group 1 had congenital or prelingual onset of subjective hearing loss, whereas most patients in the other groups (except three patients in group 2) had postlingual onset or normal hearing. Moreover, patients in group 1 were younger at ascertainment than patients in groups 2 and 3, although this difference was not statistically significant (Fig. 2d, p = 0.132 by Kruskal–Wallis rank sum test). In summary, our audiological data suggest that truncated alleles are associated with more severe phenotypes for both the extent and severity of progressive hearing loss; these effects were greater for biallelic truncated alleles, implying that the USH2A gene exhibits an allelic hierarchy from an audiological perspective.

Hearing thresholds and ages at ascertainment, stratified by group. (a) Hearing thresholds, stratified by group (1 vs. 2 vs. 3). (b) Hearing thresholds, stratified by group (1 + 2 vs. 3). (c) Hearing thresholds, stratified by group (2 vs. 3). (d) Age at ascertainment, stratified by group (1 vs. 2 vs. 3).
Retinal phenotypes and allelic hierarchy
As previously reported, age has a strong effect on retinal phenotypes. Patients under 16 years of age did not report ophthalmological symptoms prior to clinical evaluation. Previous report shows that the onset of syndromic RP, although earlier than non-syndromic RP, is still delayed after adolescence15,16,17. Eight of the patients were under 16 years old, and their phenotypes mimicked nonsyndromic hearing loss. Therefore, eight patients aged < 16 years were excluded from analyses of retinal phenotypes; this led to exclusion of all participants from group 1. The remaining patients in group 2 were 33, 47, 57, and 57 years old; the remaining patients in group 3 were 30, 37, 40, and 65 years old. Visual acuity, electroretinography (ERG), Goldmann visual field (GVF), optical coherence tomography (OCT), fundus photographs, and fundus autofluorescence (FAF) assessments were used to evaluate each patient’s retinal phenotype.
The median best-corrected visual acuity values, determined using the logarithm of the minimum angle of resolution (logMAR) scale, showed that there was no significant difference in retinal function among the groups (group 2: median = 0.699, range = 0–1.699; group 3: median = 0.134, range = 0–2; Fig. 3a; p = 0.597 by Wilcoxon rank sum test). All patients included in this study did not have other ophthalmological diseases, which may affect visual acuity, such as cataract or preretinal fibrosis, at the time of evaluation. Additionally, we evaluated ERG responses for each patient. The ERG response of many patients with RP is undetectable, which indicates reduced or no function28. We measured residual responses using three different ERG settings: 0.01 scotopic ERG for a rod response, 3.0 scotopic ERG for a combined rod and cone response, and a photopic test for a cone response. The response of 0.01 scotopic ERG in all patients were undetectable. These results are consistent with the findings in previous reports, in which the progression of RP initially affected rod function13,14. Only one-fourth of the patients in group 2 exhibited residual cone function, whereas half of the patients in group 3 exhibited a cone response. Representative graphs for 3.0 scotopic ERG are shown in Fig. 3b. The 30-Hz flicker test evaluates cone function and was used to investigate RP in a previous study29. Group 3 patients exhibited higher response amplitudes compared with group 2 patients, indicating that cone function had been more effectively preserved in group 3 patients, but it was statistically insignificant (p = 0.215 by Wilcoxon rank sum test; Fig. 3c). As ERG is often altered in RP patients before other signs or symptoms appear, the ERG of excluded patients under age 16 were also analyzed. With the exception of one patient in group 2, 0.01 scotopic ERG response was undetectable. The photopic ERG response was found in all excluded patients due to age, indicating the preservation of cone function in younger ages. The visual fields of all patients in group 2 and two patients in group 3 were restricted to a central area. However, the other two patients in group 3 had relatively preserved visual fields (Fig. 3d). These ERG and GVF results reveal that patients with USH2A-related RP who have truncated alleles tend to exhibit more severe functional retinal degeneration.

Comparison of functional consequences of retinal degeneration between patients of Groups 2 and 3. (a) Visual acuities in logMAR scale in Group 2 and Group 3 patients. (b) Representative scotopic 3.0 electroretinograms of Group 2 (above) and Group 3 (below) patients. Each column of the x-axis represents 25 ms, and each column of the y-axis represents 100 mV. (c) Response amplitudes in ERG 30 Hz flicker tests in Group 2 and Group 3 patients. (d) Maximal isopters in Goldmann perimetry in Group 2 and Group 3 patients. logMAR, logarithm of the minimum angle of resolution; ERG, electroretinography.
For evaluation of retinal structure, OCT was used to examine the integrity of the junctional complex, which consists of an interdigitation zone, an ellipsoid zone, and an external limiting membrane30. Only one-fourth of the patients in group 2 exhibited conserved junctional complexes, compared with three-fourths of the patients in group 3 (Fig. 4a). Additionally, we compared retinas from patients in groups 2 and 3 with respect to bony spicules and waxy optic discs, which indicate retinal degeneration31. Fundus photographs revealed that group 2 patients had more bony spicules, and these were distributed more widely. Although this difference may occur from the difference of duration of the disease, the number and distribution, combined with overall retinal intactness, supports that group 2 patients show more severe phenotypes in a qualitative manner. In addition, the spicules in group 2 patients were thicker and wider in size and more apparent overall. Furthermore, all four patients in group 2 exhibited waxy optic discs, indicating progressing pallor; only one-fourth of the patients in group 3 exhibited similarly degenerate optic discs (Fig. 4b). Patients in group 2 also exhibited more paramacular mottling in FAF images than patients in group 3. Patients from both groups also underwent evaluation of hyper-autofluorescent rings (i.e., Robson–Holder rings), which are characteristic of RP32,33. The ring structures present in group 3 patients were less obvious than the structures present in group 2 patients or were completely absent. In addition, although being fainter, the integrity of the ring structure was more intact in group 2 patients, indicating the absence of the hyper-autofluorescent ring is not due to further advance of degeneration. Indicating less severe phenotypes in group 3 (Fig. 4c). Overall, these OCT, fundus, and FAF results indicate that patients with truncated alleles tend to exhibit more severe structural disruption in their retinas.

Comparison of structural consequences between patients of Groups 2 and 3. (a) Representative images of optical coherence tomography of Group 2 (left) and Group 3 (right) patients. (b) Representative wide fundus photographs of Group 2 (left) and Group 3 (right) patients. The arrows indicate bony spicules, an indication of retinal degeneration. (c) Representative image of fundus autofluorescence of Group 2 (left) and Group 3 (right) patients. The asterisk (*) indicates the hyperfluorescent fovea and arrows indicate hypo-fluorescent mottles in the paramacular region.
Correlations between audiological and ophthalmological phenotypes
In this study, we analyzed the correlation between audiological and ophthalmological phenotypes in eight suitable patients (groups 2 and 3; Fig. 5). Although mean hearing threshold was not correlated with logMAR visual acuity (Spearman correlation coefficient = 0.137, p = 0.613), 30 Hz flicker ERG amplitude (Spearman correlation coefficient = − 0.456, p = 0.076), or the maximum isopter on GVF evaluation (Spearman correlation coefficient = 0.050, p = 0.853), there was a notable correlation between mean hearing threshold and 30-Hz flicker ERG implicit time (Spearman correlation coefficient = − 0.525, p = 0.037). In particular, the correlation had distinct effects on the presence of a truncated allele. Group 2 patients exhibited a negative correlation (Spearman correlation coefficient = − 0.775, p = 0.024), whereas group 3 patients exhibited no discernible correlation (Spearman correlation coefficient = − 0.271, p = 0.515). In the same vein, subgroup analysis of Group 2 revealed correlations between hearing threshold and visual acuity (Spearman correlation coefficient = 0.859, p = 0.006) and between hearing threshold and 30 Hz flicker ERG amplitude (Spearman correlation coefficient = − 0.827, p = 0.011).

Relationships between hearing threshold and ophthalmological phenotypes. (a) Hearing threshold and logMAR visual acuity. (b) Hearing threshold and 30-Hz flicker ERG amplitude. (c) Hearing threshold and 30-Hz flicker ERG implicit time. (d) Hearing threshold and the maximum isopter produced by Goldmann visual field evaluation. Groups 2 and 3 values are represented by white (○) and black circles (●), respectively.
Discussion
This study provides a comprehensive analysis of the clinical features of USH2A-related disorders and presents evidence for an allelic hierarchy in South Korean patients. Our results show that patients with at least one truncated variant exhibit more severe audiological and retinal phenotypes, compared with patients who have biallelic nontruncated variants. Notably, this trend is even greater in patients with biallelic truncated variants, suggesting that the severity of disorders is determined by the presence and number of truncated mutant alleles. Additionally, there was a clear correlation between mean hearing threshold and 30-Hz flicker ERG implicit time, depending on the presence of a truncated allele. These findings have important clinical implications because data regarding genotype–phenotype correlations and allelic hierarchies can be used to predict the natural course of disease and to guide the use of gene therapies.
Several medical genetic syndromes involve allelic hierarchies, including cystic fibrosis, hemophilia, neurofibromatosis type I, and Usher syndrome12,34,35,36. Phenotype severity depends on the particular mutant allele and its effect on protein function. The USH2A gene exhibits an allelic hierarchy; there are tissue-specific genotype–phenotype correlations for various clinical manifestations of USH2A-related disorders, including hearing loss, RP, and olfactory function37,38,39. The results of previous studies suggested an allelic hierarchy for nonsyndromic RP that was associated with the USH2A gene, particularly for the allelic variant p.Cys759Phe. Those studies showed that patients who were heterozygous for p.Cys759Phe and had an additional truncated or USH2-related missense variant tended to exhibit earlier onset and more severe clinical phenotypes, compared with patients who were homozygous for p.Cys759Phe19. Furthermore, truncated alleles of USH2A are more likely to have deleterious effects on protein structure and function; they may result in more severe clinical phenotypes, compared with missense or other nontruncated variants. These phenomena occur because truncated variants often lead to premature protein termination, resulting in a loss of function or a mutant protein that cannot fulfill its normal physiological role40. Recent allelic hierarchy data suggest that the presence of biallelic truncated alleles is likely to cause severe audiological phenotypes, such as hearing loss. For example, homozygous truncated c.2299delG allelic variants are associated with severe and progressive hearing loss in patients with USH2A-related disorders21. Findings in our previous study suggested that a particular combination of biallelic truncated alleles of USH2A could result in more severe audiological phenotypes and a greater need for CI, compared with other allele combinations12. The present study provides further support for this observation because we identified a patient (SH351) with two truncated alleles who exhibited severe-to-profound progressive hearing loss and who underwent CI before the age of 13 years. Therefore, an understanding of the potential impacts of allelic hierarchies on audiological phenotypes is important for personalized audiological rehabilitation. The results of this study improve the broader understanding of allelic hierarchies from an audiological perspective; they reveal that the presence of at least one truncated allele can result in earlier onset of subjective hearing loss and more severe thresholds. We also noted greater effects for biallelic truncated alleles, an observation that is consistent with the dose-dependent effect on hearing loss severity identified in the RUSH2A study37. Furthermore, the allelic hierarchy was not restricted to one particular truncated allele, indicating that different truncated variants of USH2A may have similar effects on audiological phenotypes. However, the present study did not evaluate missense alleles in the N-terminal laminin domain of USH2A; two of these alleles are associated with USH2 but not with nonsyndromic RP. Therefore, compared with truncated alleles, the presence of these missense alleles may have different effects on clinical presentation41.
For the 10 patients identified with non-congenital hearing loss, we made an analysis to determine the rate of auditory decline since birth (i.e., hearing loss progression, dB/year). We then assessed the correlation coefficient between the hearing loss progression and subjective onset of hearing loss (prelingual, postlingual, and normal). This analysis yielded a Spearman’s rank correlation coefficient of − 0.669 (p = 0.035), suggesting a inverse relationship. In other words, this finding poses that an accelerated rate of hearing loss progression is associated with an earlier onset of subjective hearing loss.
Our ophthalmological findings also suggest that the USH2A gene exhibits an allelic hierarchy. All ophthalmological examinations revealed differences in the severity of RP symptoms between patients with and without truncated alleles. Functional measurements and structural studies yielded similar results, showing that patients with truncated alleles exhibited accelerated structural degeneration leading to loss of visual function, as previously reported42. The confounding effects of age on RP severity have been reported for patients with various genetic backgrounds, regardless of specific mutations15,16. However, an older patient from group 3 (SH637) exhibited superior functional and structural preservation compared with a younger patient from group 2 (SH608). This example suggests that the genetic influence of USH2A is sufficiently strong to control the natural progression of RP. One patient from group 3 (SH707) exhibited a particularly severe retinal phenotype, with collapsed retinal structure and significant losses affecting both visual field and visual acuity. This patient had the previously unreported c.4372C > T variant, which clearly has severe effects on retinal structure and function. Consequently, there are exceptions to the allelic hierarchy, such that some nontruncated variants elicit severe phenotypes. Nontruncated variants reportedly have various levels of pathogenicity in USH2A-related Usher syndrome. A more elaborate hierarchical system based on both truncation number and nontruncated variant pathogenicity may provide a better understanding of the relationship between genotypes and phenotypes.
Importantly, RP is strongly associated with age15,16. Even patients with pathogenic gene variants may not notice the onset of RP for an extended interval. Therefore, the lack of correlation between auditory and visual phenotypes may be related to differences in the onset and progression of these phenotypes. Despite a few exceptions, the allelic hierarchy between groups was clearly based on the presence of truncating alleles. Future studies, including long-term follow-up of younger patients with Usher syndrome, may provide further insights regarding auditory–visual correlations and the confounding effects of age.
An understanding of allelic hierarchies may enable clinicians to predict the course of clinical phenotypes associated with USH2A-related disorders. This information may also be used to develop personalized monitoring plans and treatment strategies tailored to the truncated or nontruncated mutant alleles involved. Therefore, early identification of USH2A biallelic variants is particularly important for patients with these disorders; this information should facilitate timely interventions and may improve outcomes10. Newborn infant screening approaches are crucial for the identification of SNHL and the underlying genetic causes of hearing loss. Screening facilitates early rehabilitation of affected individuals. Our studies show that retinal phenotypes may not be present when hearing loss is first observed in pediatric patients with USH2, even if the patient has two truncated USH2A alleles. Therefore, USH2A-related USH2 can mimic nonsyndromic hearing loss. Nonetheless, the allelic hierarchy we propose suggests that group 3 patients may experience auditory deterioration over time, eventually requiring CI. This deterioration may be accompanied by the appearance of retinal phenotypes that are more severe than expected43. Furthermore, therapeutic strategies in development for USH2A-related disorders44,45,46 include gene replacement, gene editing, and antisense oligonucleotide-based treatment. Clinical trials for USH2A-related RP using gene therapy are currently underway47, and the efficacy of gene therapy on hearing loss has been demonstrated in preclinical studies48. Here, we used WGS to identify two deep intronic variants that introduce cryptic splicing. These variants may be good candidates for therapy using antisense oligonucleotides, which prevent aberrant splicing and potentially rescue USH2A-related phenotypes. Notably, the use of allelic hierarchy data to predict clinical phenotypes may indirectly identify optimal therapeutic windows for gene therapy and other targeted treatments. An understanding of the impact of specific USH2A allele combinations on clinical presentation can guide the development of personalized treatment strategies and improve outcomes for affected individuals.
This study had some limitations that should be addressed in future studies. Our sample size, comprising 16 participants, was relatively small, thereby limiting the generalizability of our findings, both within the context of the South Korean population and beyond, particularly concerning individuals with USH2A variants. Nonetheless, to derive and augment such understanding, a meticulous analysis of clinical data and genotypes is imperative, even amidst the challenges posed by these rare cases. Future studies with larger sample sizes and more diverse patient populations are needed to validate and expand upon our findings. Additionally, our study lacked longitudinal analyses, and we did not elucidate how the USH2A-based allelic hierarchy operates over time. Future studies that include longitudinal follow-up and analyses of disease progression are needed to better understand the natural course of clinical phenotypes associated with USH2A-related disorders, as well as the impacts of particular genetic variants on disease progression. To assess potential progression, it is crucial to account for the concurrent presence of presbycusis or the gradual decline in lip-reading skills associated with visual deterioration. In our cohort, four participants aged 40 or above were identified as susceptible to presbycusis. Unfortunately, speech assessments were not administered to these individuals, precluding an evaluation of lip-reading. This is another limitation of our study. Despite these limitations, this study provides important evidence for an USH2A-related allelic hierarchy in South Korean patients. Our observations will lead to a better understanding of the natural course of clinical phenotypes and will improve the use of genotype-based therapy in the current era of precision medicine.
Methods
Study participants
The study protocol was approved by the Institutional Review Board of Seoul National University Hospital (IRB-H-0905-041-281, and IRB-H-2202-045-1298). Written informed consent was obtained from the participants or their legal guardians, in accordance with the tenets of the Declaration of Helsinki. A retrospective review was performed using the in-house database for genetic hearing loss at Seoul National University Hospital. Genomic DNA from 419 probands was subjected to clinical exome sequencing or WGS, regardless of audiological phenotypes or inheritance patterns. Probands with USH2A biallelic variants, either compound heterozygotes or homozygotes, were included. In total, 16 participants (9 male individuals and 7 female individuals) from 14 unrelated South Korean families with USH2A biallelic variants (3.3%) were identified. The mean age at ascertainment was 25.5 years (range, 0.3–64 years). Demographics, clinical phenotypes, and genotypes were evaluated.
Audiological phenotyping
We analyzed audiological phenotypes in terms of the following four parameters: degree of severity, onset of hearing impairment, symmetry of hearing loss, and progression observed during follow-up evaluations. Pure-tone audiometry was used to assess the hearing thresholds of participants at six different octave frequencies (0.25, 0.5, 1, 2, 4, and 8 kHz), considering the participant’s age. For participants who were aged 90 dB), based on the mean thresholds. Subjective onset of hearing loss information was obtained from each participant’s medical records or determined using the date of the first audiogram. Hearing asymmetry was defined as a difference of ≥ 15 dB between the mean hearing thresholds for both ears at 500, 1000, 2000, and 4000 Hz on pure-tone audiometry; alternatively, it was determined using the auditory steady-state response.
Ophthalmological phenotyping
Clinical ophthalmic examinations included best-corrected visual acuity, ERG, GVF, OCT, fundus photography, and FAF. All examinations were preceded by pupil dilation. ERG was conducted in accordance with International Society for Clinical Electrophysiology of Vision standards using a commercial ERG system (Retiport32; Roland Consult, Brandenburg an der Havel, Germany)49. GVF evaluations were performed by professionals, in accordance with standard protocols50,51. OCT data were obtained using the Heidelberg Spectralis OCT instrument (Heidelberg Engineering, Hemel Hempstead, UK). Fundus photographs and FAF data were obtained using the Optos retinal imaging system (Optos, Marlborough, MA, USA).
Whole-exome sequencing
Genomic DNA extracted from peripheral blood samples were collected from probands and subjected to exome sequencing using the Sure Select 50 Mb Hybridization and Capture kit (Agilent Technologies, Santa Clara, CA, USA). A library was prepared following the manufacturer’s instructions and was paired-end sequenced using a NovaSeq 6000 sequencing system (Illumina, San Diego, CA, USA) with an average coverage depth of 100×. The paired-end read length was 100 bp, and sequence reads were aligned with the University of California Santa Cruz hg19 reference genome browser (https://genome.ucsc.edu/). Subsequent to this, data were processed using the Genome Analysis Toolkit (GATK), adhering to the best-practice pipeline for the identification of Single Nucleotide Variants (SNVs) and insertions/deletions (indels)52. Variant annotation was executed using the ANNOVAR program, leveraging sources such as the RefSeq gene set and gnomAD53,54. Candidate variants were selectively identified, prioritizing rare non-silent variants, which encompassed nonsynonymous SNVs, coding indels, and splicing variants. For additional filtration of ethnic-specific variants, Korea Reference Genome Database (KRGDB) and Korean Variant Archive (KOVA) databases were used55,56. Additionally, the Clinvar and HGMD databases were screened to ascertain whether or not candidate variants have been previously detected in other families57,58. As previously described12,59,60,61,62,63,64, thorough bioinformatics analysis was conducted with strict filtering to identify candidate variants associated with SNHL and/or RP. For phasing analysis, segregation of these variants was investigated via Sanger sequencing of parental DNA samples.
Whole-genome sequencing
DNA libraries for WGS were generated using the TruSeq DNA PCR-Free kit (Illumina) from 1 µg of genomic DNA. WGS was conducted on the NovaSeq platform 6000 (Illumina) to generate 151-bp paired-end reads with an average coverage depth of 30×. Sequenced reads were mapped to the human reference genome (GRCh38) using the BWA-MEME algorithm65,66, and duplicated reads were removed using SAMBLASTER67. Base substitutions and short indels were identified using HaplotypeCaller52 and trelka268. Variants identified by either of these tools were included in further analyses. Genomic rearrangements were identified using Delly69; the breakpoints of genomic rearrangements of interest were visually inspected and confirmed using Integrative Genomics Viewer70. For phasing analysis, segregation of these variants was investigated via Sanger sequencing of parental DNA samples.
Statistical analyses
All analyses were performed using R software (ver. 4.2.2; R Development Core Team, Vienna, Austria). The age at onset of subjective hearing loss, age at ascertainment, and degree of hearing loss were identified for the different genotype groups. Hearing thresholds were determined for the different frequencies, and mean values were calculated for each group. The Kruskal–Wallis rank sum test was used to evaluate differences in hearing threshold at each frequency among the three groups. For each frequency, each participant’s mean hearing threshold from both ears was analyzed. Correlations between audiological and ophthalmic phenotypes were evaluated using participants whose ophthalmic phenotypes had been investigated. Visual acuity, ERG, and GVF evaluations were used to investigate ophthalmic phenotypes. The pure-tone hearing threshold was used to investigate audiological phenotypes. Correlations between ophthalmic test values and hearing threshold values were evaluated using Spearman rank correlation analysis.
Data availability
Sequence variations were submitted to Clinvar with the accession code (SCV003844066-SCV003844069) https://www.ncbi.nlm.nih.gov/clinvar/submitters/508922. The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The data (Individual-level whole exome/genome sequencing) that support the findings of this study are available from the corresponding author (S-Y.L, maru4843@hanamil.net) upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
References
-
Nisenbaum, E. et al. Review of genotype–phenotype correlations in Usher syndrome. Ear Hear. 43, 1–8 (2022).
Google Scholar
-
de Guimaraes, T. A. C., Arram, E., Shakarchi, A. F., Georgiou, M. & Michaelides, M. Inherited causes of combined vision and hearing loss: Clinical features and molecular genetics. Br. J. Ophthalmol. 107, 1403 (2022).
Google Scholar
-
Bahena, P. et al. Unraveling the genetic complexities of combined retinal dystrophy and hearing impairment. Hum. Genet. 141, 785–803 (2022).
Google Scholar
-
Fuster-García, C. et al. Usher syndrome: Genetics of a human ciliopathy. Int. J. Mol. Sci. 22, 6723 (2021).
Google Scholar
-
Tebbe, L. et al. The usherin mutation c. 2299delG leads to its mislocalization and disrupts interactions with whirlin and VLGR1. Nat. Commun. 14, 972 (2023).
Google Scholar
-
Stemerdink, M. et al. Genetics, pathogenesis and therapeutic developments for Usher syndrome type 2. Hum. Genet. 141, 1–22 (2021).
-
Toms, M., Pagarkar, W. & Moosajee, M. Usher syndrome: Clinical features, molecular genetics and advancing therapeutics. Ther. Adv. Ophthalmol. 12, 2515841420952194 (2020).
Google Scholar
-
Liu, X. et al. Generation and genetic correction of USH2A c. 2299delG mutation in patient-derived induced pluripotent stem cells. Genes 12, 805 (2021).
Google Scholar
-
Molina-Ramírez, L. P. et al. Establishing genotype–phenotype correlation in USH2A-related disorders to personalize audiological surveillance and rehabilitation. Otol. Neurotol. 41, 431–437 (2020).
Google Scholar
-
Brodie, K. D. et al. Genetic testing leading to early identification of childhood ocular manifestations of usher syndrome. Laryngoscope 131, E2053–E2059 (2021).
Google Scholar
-
Sadeghi, A. M., Cohn, E. S., Kimberling, W. J., Halvarsson, G. & Möller, C. Expressivity of hearing loss in cases with Usher syndrome type IIA. Int. J. Audiol. 52, 832–837 (2013).
Google Scholar
-
Lee, S.-Y. et al. Severe or profound sensorineural hearing loss caused by novel USH2A variants in Korea: Potential genotype–phenotype correlation. Clin. Exp. Otorhinolaryngol. 13, 113–122 (2020).
Google Scholar
-
Sengillo, J. D. et al. Electroretinography reveals difference in cone function between syndromic and nonsyndromic USH2A patients. Sci. Rep. 7, 11170. https://doi.org/10.1038/s41598-017-11679-y (2017).
Google Scholar
-
Jauregui, R. et al. Disease asymmetry and hyperautofluorescent ring shape in retinitis pigmentosa patients. Sci. Rep. 10, 3364. https://doi.org/10.1038/s41598-020-60137-9 (2020).
Google Scholar
-
Tsujikawa, M. et al. Age at onset curves of retinitis pigmentosa. Arch Ophthalmol. 126, 337–340. https://doi.org/10.1001/archopht.126.3.337 (2008).
Google Scholar
-
Cross, N., van Steen, C., Zegaoui, Y., Satherley, A. & Angelillo, L. Retinitis pigmentosa: Burden of disease and current unmet needs. Clin. Ophthalmol. 16, 1993–2010. https://doi.org/10.2147/OPTH.S365486 (2022).
Google Scholar
-
Colombo, L. et al. Spectral-domain optical coherence tomography analysis in syndromic and nonsyndromic forms of retinitis pigmentosa due to USH2A genetic variants. Ophthalmic Res. 65, 180–195. https://doi.org/10.1159/000520329 (2022).
Google Scholar
-
Perrino, P. A. et al. Multi-level evidence of an allelic hierarchy of USH2A variants in hearing, auditory processing and speech/language outcomes. Commun. Biol. 3, 180 (2020).
Google Scholar
-
Lenassi, E. et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 23, 1318–1327 (2015).
Google Scholar
-
Inaba, A. et al. Truncating variants contribute to hearing loss and severe retinopathy in USH2A-associated retinitis pigmentosa in Japanese patients. Int. J. Mol. Sci. 21, 7817 (2020).
Google Scholar
-
Hartel, B. P. et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear. Res. 339, 60–68 (2016).
Google Scholar
-
Spielman, R. S. et al. Common genetic variants account for differences in gene expression among ethnic groups. Nat. Genet. 39, 226–231 (2007).
Google Scholar
-
Li, W. et al. Genetic characteristics and variation spectrum of USH2A-related retinitis pigmentosa and usher syndrome. Front. Genet. 13, 900548. https://doi.org/10.3389/fgene.2022.900548 (2022).
Google Scholar
-
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613 (2018).
Google Scholar
-
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Google Scholar
-
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Google Scholar
-
Baux, D. et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 7, 16783 (2017).
Google Scholar
-
Shen, C. et al. Whole-exome sequencing identified genes known to be responsible for retinitis pigmentosa in 28 Chinese families. Mol. Vis. 28, 96–113 (2022).
Google Scholar
-
Creel, D. J. Electroretinograms. Handb. Clin. Neurol. 160, 481–493. https://doi.org/10.1016/B978-0-444-64032-1.00032-1 (2019).
Google Scholar
-
Messner, A. et al. Quantification of intrinsic optical signals in the outer human retina using optical coherence tomography. Ann. N. Y. Acad. Sci. 1510, 145–157. https://doi.org/10.1111/nyas.14721 (2022).
Google Scholar
-
Meng, X., Liu, X., Li, Y., Guo, T. & Yang, L. Correlation between genotype and phenotype in 69 Chinese patients with USH2A mutations: A comparative study of the patients with Usher Syndrome and Nonsyndromic Retinitis Pigmentosa. Acta Ophthalmol. 99, e447–e460. https://doi.org/10.1111/aos.14626 (2021).
Google Scholar
-
Pichi, F., Abboud, E. B., Ghazi, N. G. & Khan, A. O. Fundus autofluorescence imaging in hereditary retinal diseases. Acta Ophthalmol. 96, e549–e561. https://doi.org/10.1111/aos.13602 (2018).
Google Scholar
-
Pole, C. & Ameri, H. Fundus autofluorescence and clinical applications. J. Ophthalmic Vis. Res. 16, 432–461. https://doi.org/10.18502/jovr.v16i3.9439 (2021).
Google Scholar
-
Santagostino, E. et al. Severe hemophilia with mild bleeding phenotype: Molecular characterization and global coagulation profile. J. Thromb. Haemost. 8, 737–743. https://doi.org/10.1111/j.1538-7836.2010.03767.x (2010).
Google Scholar
-
Jett, K. & Friedman, J. M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 12, 1–11. https://doi.org/10.1097/GIM.0b013e3181bf15e3 (2010).
Google Scholar
-
Basaran, A. E. et al. Association between cystic fibrosis severity markers and CFTR genotypes in Turkish children. Turk. Thorac. J. 22, 426–431. https://doi.org/10.5152/TurkThoracJ.2021.20282 (2021).
Google Scholar
-
Hufnagel, R. B. et al. Tissue-specific genotype–phenotype correlations among USH2A-related disorders in the RUSH2A study. Hum. Mutat. 43, 613–624 (2022).
Google Scholar
-
Iannaccone, A. et al. Auditory and olfactory findings in patients with USH2A-related retinal degeneration—Findings at baseline from the rate of progression in USH2A-related retinal degeneration natural history study (RUSH2A). Am. J. Med. Genet. Part A 185, 3717–3727 (2021).
Google Scholar
-
Lad, E. M. et al. Baseline Microperimetry and OCT in the RUSH2A study: Structure−function association and correlation with disease severity. Am. J. Ophthalmol. 244, 98–116 (2022).
Google Scholar
-
Zhu, T. et al. USH2A variants in Chinese patients with Usher syndrome type II and non-syndromic retinitis pigmentosa. Br. J. Ophthalmol. 105, 694–703 (2021).
Google Scholar
-
Gao, F.-J. et al. Prevalence and genetic–phenotypic characteristics of patients with USH2A mutations in a large cohort of Chinese patients with inherited retinal disease. Br. J. Ophthalmol. 105, 87–92 (2021).
Google Scholar
-
Toualbi, L., Toms, M. & Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 201, 108330. https://doi.org/10.1016/j.exer.2020.108330 (2020).
Google Scholar
-
Toms, M. et al. Clinical and preclinical therapeutic outcome metrics for USH2A-related disease. Hum. Mol. Genet. 29, 1882–1899 (2020).
Google Scholar
-
Major, L., McClements, M. E. & MacLaren, R. E. New CRISPR tools to correct pathogenic mutations in usher syndrome. Int. J. Mol. Sci. 23, 11669 (2022).
Google Scholar
-
Zaw, K. et al. Pathogenesis and treatment of usher syndrome type IIA. Asia-Pac. J. Ophthalmol. 11, 369–379 (2022).
Google Scholar
-
Delmaghani, S. & El-Amraoui, A. The genetic and phenotypic landscapes of Usher syndrome: From disease mechanisms to a new classification. Hum. Genet. 141, 709–735 (2022).
Google Scholar
-
Dulla, K. et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Therapy 29, 2441–2455 (2021).
Google Scholar
-
Pendse, N. D. et al. in Retinal Degenerative Diseases: Mechanisms and Experimental Therapy. 91–96 (Springer).
-
Robson, A. G. et al. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 144, 165–177. https://doi.org/10.1007/s10633-022-09872-0 (2022).
Google Scholar
-
Wong, S. H. & Plant, G. T. How to interpret visual fields. Pract. Neurol. 15, 374–381. https://doi.org/10.1136/practneurol-2015-001155 (2015).
Google Scholar
-
Spector, R. H. in Clinical Methods: The History, Physical, and Laboratory Examinations (eds H. K. Walker, W. D. Hall, & J. W. Hurst) (1990).
-
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11. https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Google Scholar
-
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. https://doi.org/10.1093/nar/gkq603 (2010).
Google Scholar
-
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7 (2020).
Google Scholar
-
Jung, K. S. et al. KRGDB: The large-scale variant database of 1722 Koreans based on whole genome sequencing. Database https://doi.org/10.1093/database/baaa030 (2020).
Google Scholar
-
Lee, J. et al. A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population. Exp. Mol. Med. 54, 1862–1871. https://doi.org/10.1038/s12276-022-00871-4 (2022).
Google Scholar
-
Stenson, P. D. et al. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. https://doi.org/10.1007/s00439-017-1779-6 (2017).
Google Scholar
-
Landrum, M. J. et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. https://doi.org/10.1093/nar/gkx1153 (2018).
Google Scholar
-
Lee, S. Y. et al. Novel molecular genetic etiology of asymmetric hearing loss: Autosomal-dominant LMX1A variants. Ear Hear. 43, 1698–1707. https://doi.org/10.1097/AUD.0000000000001237 (2022).
Google Scholar
-
Lee, S. Y. et al. Identification of a potential founder effect of a novel PDZD7 variant involved in moderate-to-severe sensorineural hearing loss in Koreans. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20174174 (2019).
Google Scholar
-
Ozieblo, D. et al. Update on CD164 and LMX1A genes to strengthen their causative role in autosomal dominant hearing loss. Hum. Genet. 141, 445–453. https://doi.org/10.1007/s00439-022-02443-y (2022).
Google Scholar
-
Lee, S.-Y. et al. Ramifications of POU4F3 variants associated with autosomal dominant hearing loss in various molecular aspects. Sci. Rep. 13, 12584 (2023).
Google Scholar
-
Lee, S.-Y. et al. Novel KCNQ4 variants in different functional domains confer genotype-and mechanism-based therapeutics in patients with nonsyndromic hearing loss. Exp. Mol. Med. 53, 1192–1204 (2021).
Google Scholar
-
Lee, S. et al. Phenotypic and molecular basis of SIX1 variants linked to non-syndromic deafness and atypical branchio-otic syndrome in South Korea. Sci. Rep. 13, 11776 (2023).
Google Scholar
-
Levin, M. G. et al. Genome-wide association and multi-trait analyses characterize the common genetic architecture of heart failure. Nat. Commun. 13, 6914. https://doi.org/10.1038/s41467-022-34216-6 (2022).
Google Scholar
-
Jung, Y. & Han, D. BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics 38, 2404–2413. https://doi.org/10.1093/bioinformatics/btac137 (2022).
Google Scholar
-
Faust, G. G. & Hall, I. M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. https://doi.org/10.1093/bioinformatics/btu314 (2014).
Google Scholar
-
Kim, S. et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Google Scholar
-
Rausch, T. et al. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. https://doi.org/10.1093/bioinformatics/bts378 (2012).
Google Scholar
-
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192. https://doi.org/10.1093/bib/bbs017 (2013).
Google Scholar
Acknowledgements
This research was supported and funded by SNUH Kun-hee Lee Child Cancer & Rare Disease Project, Republic of Korea (Grant Number: FP-2022-00001-004 to Sang-Yeon Lee). This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Education [Grant Number: 2022R1C1C1003147 to Sang-Yeon Lee]. This study was supported by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (NRF-2023M3A9I4009901) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2022R1A2C1003768). This study was supported by the Genome Editing Research Program funded from the Korean government (MSIT) (RS-2023-00260351).
Author information
Authors and Affiliations
Contributions
S-Y.L. performed genomic and bioinformatic analysis. D.W.N. and Y.K.S. performed data analysis and wrote the manuscript. J.H.K., E.K.L., K.H.P., J.H.C., B.Y.C., J.H.L., and S.H.O. contributed to data collection and manuscript preparation. D.H.J. and S-Y.L. wrote the manuscript, and contributed to the study design. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Figure S1.
Supplementary Table 1.
Supplementary Table 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Nam, D.W., Song, Y.K., Kim, J.H. et al. Allelic hierarchy for USH2A influences auditory and visual phenotypes in South Korean patients.
Sci Rep 13, 20239 (2023). https://doi.org/10.1038/s41598-023-47166-w
-
Received: 14 March 2023
-
Accepted: 09 November 2023
-
Published: 19 November 2023
-
DOI: https://doi.org/10.1038/s41598-023-47166-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.