
Blood
UK Approves Groundbreaking CRISPR-Based Gene Therapy For Sickle Cell Disease And Thalassemia
Nov
This week marks an incredible win for modern medicine. The first CRISPR-based gene therapy has just been approved for clinical use in the United Kingdom. Casegvy (Exa-cel), developed by Vertex and CRISPR Therapeutics, relies on a precise gene-cutting technology first published in 2012. This technology won scientists Jennifer Doudna and Emmanuelle Charpentier the 2020 Nobel Prize in Chemistry. Now, a mere decade since its initial publication, this innovation has the potential to impact people’s lives—albeit under certain circumstances.
A Novel Therapy for Inherited Blood Diseases
Exa-cel is the first CRISPR-based gene therapy to receive regulatory approval in the world. The product is authorized to treat eligible patients ages 12 and older with either sickle cell disease or thalassemia.
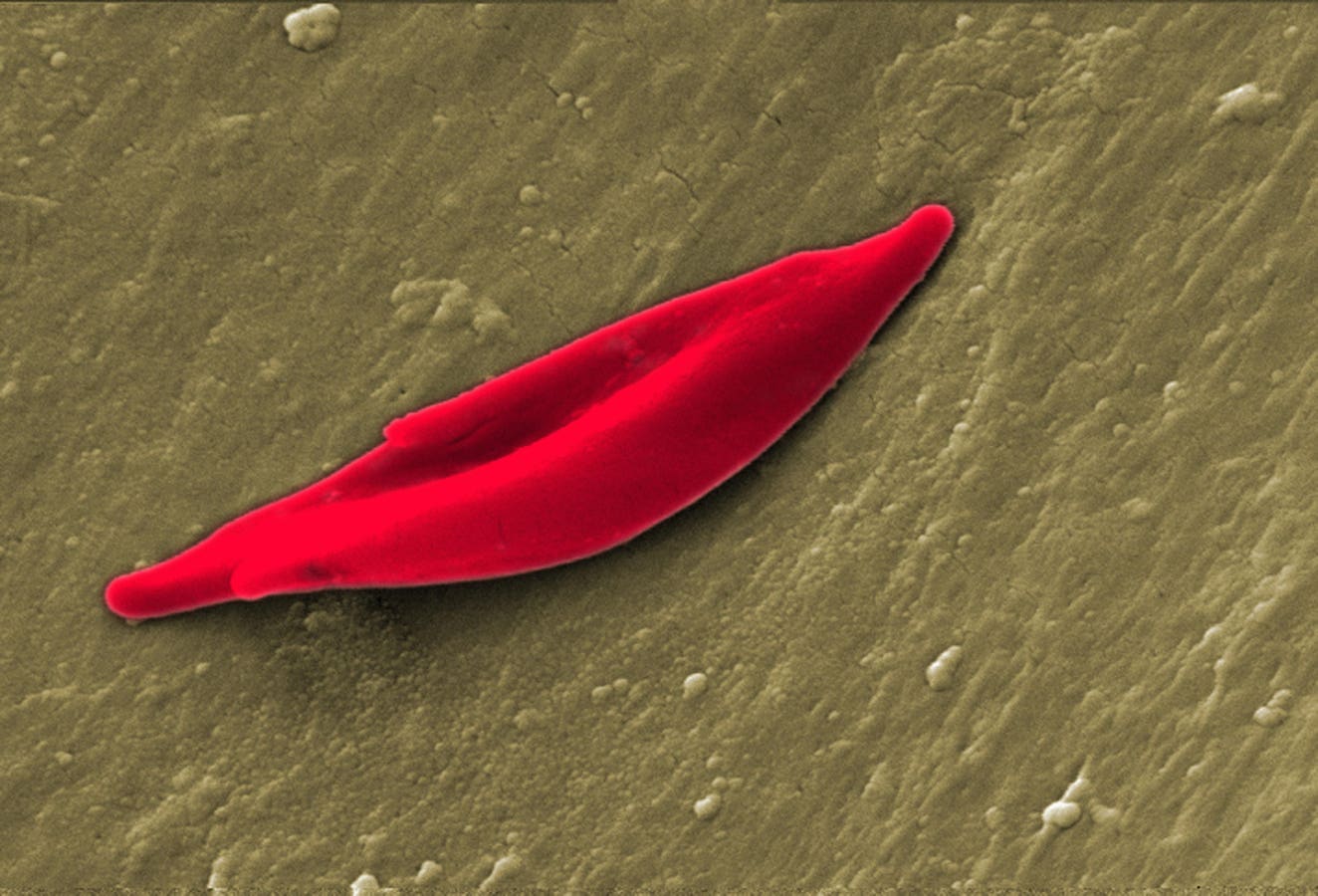
Sickle cell disease is the most common genetic blood disorder in the United States. It affects around 100,000 Americans each year, with a higher prevalence in African Americans. The condition contorts the healthy shape of red blood cells into a crescent or “sickle” shape. This sticky form blocks blood flow and creates further problems, such as episodes of severe pain and organ damage.
Thalassemia is a rarer illness that is also passed down from parent to child. People with the condition do not produce enough red blood cells and hemoglobin, a crucial protein that allows red blood cells to deliver oxygen throughout the body. The resulting anemia leaves individuals fatigued, pale and weak.
Long-term treatment is usually required to manage symptoms. For example, people with thalassemia often need blood transfusions to remove the diseased blood cells and replace them with healthy ones. A bone marrow transplant from a matched donor was considered the only cure for either condition. However, only a minority of patients with severe disease elect this option due to the significant risks involved (e.g., organ toxicity, engraftment injection).
Exa-cel could change this landscape. A single course of this treatment promises to free thalassemia patients of transfusions and sickle cell disease patients of pain crises for life. To understand more, let’s delve into how this intervention works.
How Exa-cel Works
The treatment begins by extracting CD34+ blood stem cells from the patient. These stem cells usually produce blood cells that worsen the patients’ disease—but not for long. The cells are then edited in the lab using CRISPR-Cas9 technology to rectify this faulty production. This step may take weeks or months to complete and ship the cells back to the hospital. Finally, patients undergo preparatory chemotherapy (busulfan) before receiving their stem cell injection. The chemotherapy wipes out any stem cells in the bone marrow and leaves space for the modified stem cells to engraft.
This product depends on CRISPR-Cas9 technology. This radical system was initially discovered in bacteria. When a virus attempts to insert its genes into bacteria, bacteria use this system to identify the invader’s DNA and selectively cut out the threat, thus preventing viral replication. CRISPR stands for “Clustered Regularly Interspaced Short Palindromic Repeats,” meaning the target DNA sequences, and Cas9 is the enzyme needed to snip the DNA.
Researchers have adapted this technology to modify genes of their choosing. Exa-cel specifically eliminates a gene called BCL11A. Removing this gene enables adults to safely produce fetal hemoglobin, a healthy form found in fetal development that typically ceases after birth.
Vertex expects this one-time modification to cure thalassemia and sickle cell disease patients permanently. Study results appear promising, but some forecasting is involved. Phase 3 clinical trials revealed that 24 out of 27 thalassemia patients who received exa-cel did not need blood transfusions for at least a year, and 16 out of 18 sickle cell disease patients did not experience pain crises for at least a year. The hope is that these stem cells will continue to proliferate without issues and eliminate symptoms for life.
Barriers and Concerns
Although exa-cel can be a revolutionary treatment for those with inherited blood diseases, lingering concerns temper the positive news. One concern voiced at the exa-cel FDA advisory meeting last month is the therapy’s off-target effects. Is it possible for the therapy to accidentally cut other genes, and what consequences would result? There are DNA sequencing tools to help researchers detect possible off-target locations, but much of how the human genome works remains a mystery; the biological ripple effects are harder to anticipate. Knowing this question remains partially unanswered, the UK decided the therapy’s benefits outweigh the risks.
Immunosuppression is another understated concern. Chemotherapy, a requirement for the therapy, weakens the immune system and leaves patients vulnerable to infections. Clinicians monitor the patient in a sterile environment for weeks after the infusion to help them recover, but some viruses may already lie within the patient. Latent viral reactivation has been documented in some bone marrow transplant patients and patients using other cell therapies such as CAR T therapy. With the immune system unbalanced, normally dormant viruses such as Epstein-barr virus (EBV), cytomegalovirus (CMV) and human herpesvirus 6 (HHV-6) can resurge and wreak havoc—this includes organ inflammation and even cancer.
Exa-cel’s impact is also limited to a minority of patients. It is not a first-line option by any means, as only individuals who cannot find a matched donor for a bone marrow transplant can qualify for the treatment; those with severe disease will need to exhaust stem cell transplant options first. An estimated 2,000 UK citizens qualify for treatment with these conditions.
Finances are another unspoken barrier. The list price for exa-cel has yet to be decided, but given the cost of other single-use gene therapies, it will likely exceed $1 million. Personalized medicines of this nature are resource and personnel-intensive, which increases expenses. Some companies insist the price tag is justified, as the intervention is meant to replace multiple years of treatments.
If left as is, this novel advance will benefit the rich alone. The ideal goal is to increase access to all who need it. Jennifer Doudna, cofounder of CRISPR-Cas9, echoed this sentiment in a recent interview, stating, “I don’t think we want to live, or I don’t want to live, in a world where only a few wealthy or connected people can get access to something like this.”
Looking Forward
Movement across the pond will soon ripple to US shores. The FDA decision dates for Exa-cel’s use for sickle cell disease and thalassemia are set for December 8th, 2023, and March 30, 2024, respectively. If approved, exa-cel will have achieved another milestone for the cell therapy field.
Exa-cel will surely be the first of many CRISPR-based gene therapies to come. Only a minority of patients will benefit from this advance for now, but possibilities continue to unfurl. Just this year, researchers published a groundbreaking paper in Science using mouse models of sickle cell disease and thalassemia, which I covered in three installments: read Part 1, Part 2 and Part 3 here. Their innovative approach involves administering a single injection that changes the cells directly inside the patient’s body. This could eliminate the need for immunosuppression and complex laboratory procedures while reducing costs significantly. Such innovations propel the field toward a bright future, poised to transform lives on a broader scale.