
Infection
Genomic analysis of Plasmodium vivax describes patterns of connectivity and putative drivers of adaptation in Ethiopia
Nov
Abstract
Ethiopia has the greatest burden of Plasmodium vivax in Africa, but little is known about the epidemiological landscape of parasites across the country. We analysed the genomic diversity of 137 P. vivax isolates collected nine Ethiopian districts from 2012 to 2016. Signatures of selection were detected by cross-country comparisons with isolates from Thailand (n = 104) and Indonesia (n = 111), representing regions with low and high chloroquine resistance respectively. 26% (35/137) of Ethiopian infections were polyclonal, and 48.5% (17/35) of these comprised highly related clones (within-host identity-by-descent > 25%), indicating frequent co-transmission and superinfection. Parasite gene flow between districts could not be explained entirely by geographic distance, with economic and cultural factors hypothesised to have an impact on connectivity. Amplification of the duffy binding protein gene (pvdbp1) was prevalent across all districts (16–75%). Cross-population haplotype homozygosity revealed positive selection in a region proximal to the putative chloroquine resistance transporter gene (pvcrt-o). An S25P variant in amino acid transporter 1 (pvaat1), whose homologue has recently been implicated in P. falciparum chloroquine resistance evolution, was prevalent in Ethiopia (96%) but not Thailand or Indonesia (35–53%). The genomic architecture in Ethiopia highlights circulating variants of potential public health concern in an endemic setting with evidence of stable transmission.
Introduction
Plasmodium vivax is becoming the predominant cause of malaria in the Asia–Pacific and Americas. However there is growing evidence of hidden reservoirs of infection within the African continent, with the highest number of cases reported from the Horn of Africa, where Ethiopia contributes 6% of the global burden of vivax malaria cases and large majority of African vivax malaria1,2. These disturbing trends highlight major shortcomings in current approaches to survey P. vivax.
P. vivax causes substantial morbidity in Ethiopia, accounting for approximately 30% of local malaria1. In 2020, the country reported a three year high of > 260 thousand cases of malaria1, and these numbers do not accurately capture the even greater burden of asymptomatic P. vivax present as blood or liver stage infections3. To date, local public health interventions in Ethiopia have been largely focused on the control of P. falciparum, but these methods have less impact on reducing the P. vivax reservoir4,5. Several biological factors make P. vivax harder to control than P. falciparum, including its ability to form dormant liver stages which can reactivate causing blood-stage infections in the absence of transmission, and the rapid development of gametocyte stages that enhance parasite transmission and complicate infection tracking. P. vivax blood-stage infections also typically have low parasite density, creating challenges in detecting and correctly diagnosing infected individuals6. In Ethiopia chloroquine remains the first line antimalarial treatment for patients with P. vivax malaria, although there are reports that its clinical efficacy may be declining7,8,9,10,11,12,13,14,15. Reliable molecular markers of chloroquine resistance (CQR) in P. vivax are urgently need to support surveillance for resistance, but these have yet to be defined16. To confound matters further there is wide heterogeneity in malaria epidemiology across Ethiopia due to significant variation in ecology, climate, altitude, and associated receptivity to Anopheline vectors. These factors further complicate public health decision-making on the most effective intervention strategies for specific regions17.
Population genomic studies of P. vivax have significant potential to deepen our understanding of the biology and epidemiology of this species. Previous genomic analyses have identified novel candidates of resistance to antimalarial drugs and selective pressures, as well as revealing a broad spectrum of diversity and population structure in different geographic regions, that reflect underlying patterns of transmission and spread of the parasite18,19,20,21,22,23,24. A recent genomic epidemiology study of 24 Ethiopian P. vivax isolates revealed a high prevalence of copy number amplification of the parasite duffy binding protein (pvdbp1) gene25; representing either a potential response to the high prevalence of duffy negativity in the human population, or an adaptive immune evasion strategy26. Using comparative analyses of extended haplotypes between Ethiopia and each of Thailand and Indonesia, the study also found evidence suggestive of selection in a region upstream of the chloroquine resistance transporter gene (pvcrt-o), a candidate determinant of chloroquine resistance25. However, the small sample size and limited geographic distribution of those samples across Ethiopia limited the inferences that could be drawn from these signals. Another recent study in Africa has identified a new malaria chloroquine resistance candidate; the amino acid transporter 1 gene (pfaat1) was implicated in the evolution of chloroquine resistance in P. falciparum27. However, the P. vivax amino acid transporter 1 gene had not been explored in Ethiopia. Here, using 137 high quality genomes collected across nine districts, we provide a detailed exploration of the natural selection and population structure of P. vivax in Ethiopia to inform on potential public health challenges.
Results
High-quality P. vivax genomes from nine districts of Ethiopia
From a total of 159 Ethiopian P. vivax genomes present in the MalariaGEN Pv4.0 dataset, we selected 137 high-quality samples (86.2%), with high-quality defined as having < 15% missing genotyping calls at a set of 410,900 high-quality biallelic SNPs (Supplementary Data 1). To compare these genomes with P. vivax from other regions, we used MalariaGEN Pv4.0 data from Thailand and Papua Indonesia25. Using the high-quality biallelic SNP set, a set of high-quality Thai (n = 104) and Indonesian (n = 111) samples were selected; a similar approach was taken in an earlier small-scale study of 24 Ethiopian P. vivax genomes25.
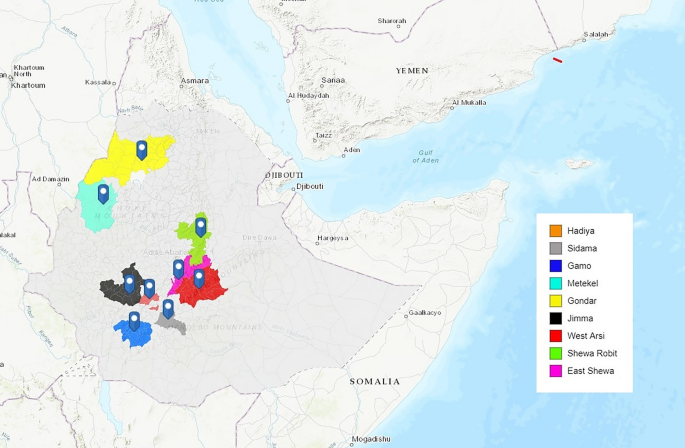
The 137 Ethiopian samples included in the analysis were obtained from 10 districts, although one district (Badowacho) contributed only 1 isolate and therefore was not included in population-level analyses. The locations of the remaining nine districts are presented in Fig. 1, with malaria endemicity data summarised in (Supplementary Table 1). Although the small sample size (n < 10 isolates) from Gondar, Metekel, East Shewa, Sidama, Hadiya and Gamo suggest caution is needed in interpreting the data from these areas (Table 1), these districts were included in analyses to enable detection of geographic trends that could be evaluated further in future studies.
Study site locations. Map illustrating the locations of the nine study sites in Ethiopia, created using ArcGIS (http://www.arcgis.com, accessed September 2022).
High frequency of superinfection and coinfection in Ethiopia
Within-sample infection complexity was assessed using the FWS score, which ranges from 0 to 1, with increasing values reflecting increasing clonality. An FWS threshold < 0.95 generally indicates polyclonal infection. Across Ethiopia, 25.5% (35/137) of infections had FWS < 0.95. At the district level, the polyclonal infection prevalence ranged from 0 to 55%, with highest prevalence of polyclonality in Gondar (Fig. 2, Table 1). Median FWS scores exceeded 0.95 in all districts aside from Gondar (median FWS = 0.92, n = 11). When all nine districts were evaluated, there was no trend between malaria endemicity stratifications based on Annual Parasite Incidence (API) and the percentage of polyclonal infections. However, the small sample size at several sites may have constrained the accuracy of these estimates. Amongst the four sites with n ≥ 10 (North Shewa, West Arsi, Jimma and Gondar), a positive trend between endemicity and polyclonality was observed.

Within-sample infection complexity by district-level administration zone. Infection complexity was determined using the within-sample F statistic (FWS). The box and whiskers reflect the median, upper and lower quartile and 95% Confidence Intervals. The plot is based on data generated using n = 137 independent infections. Shewa Robit is a health centre in North Shewa district.
Plots of the non-reference allele frequency (NRAF) distributions across the genome were created for the 35 polyclonal infections, revealing a spectrum of within-host relationships (Supplementary Fig. 1). DEploid was used to phase the genomes of the clones making up ≥ 10% of an infection. The relatedness between the clones within each polyclonal infection was then evaluated using identity-by-descent (IBD) measures. In total 42.8% (15/35) of the isolates comprised at least one pair of clones with high relatedness (≥ 25% IBD) indicative of half-siblings or greater and likely reflecting co-infection (single mosquito inoculation, co-transmission) rather than superinfection (multiple inoculations) events. At the district-level, the prevalence of putative superinfections (defined as IBD < 25%) ranged from 0 to 100% (Table 1). Amongst the four sites with ≥ 10 isolates, there was no apparent trend between endemicity and superinfection prevalence.
Subtle population structure between northern and southern regions of Ethiopia
IBD was used to capture patterns of relatedness between infections, both within and between districts. The median IBD between infections ranged from 5 to 10% across the districts, with the highest levels observed in Sidama (Table 1, Fig. 3). Connectivity plots illustrating relatedness at a spectrum of IBD thresholds (5, 7.5, 10, 25, 50 and 95%) revealed that, apart from one isolate from Jimma (SGH-1-357), all infections shared at least 5% of their genome with one other infection from across the Ethiopian data set (Fig. 3a). At the 7.5% IBD threshold, many of the infections from the northern districts of Gondar and North Shewa formed a distinct cluster from the other districts, and the isolates from Jimma revealed relatively lower connectivity relative to the other infections in the southern districts cluster (Fig. 3b). At thresholds above 10% IBD, the geographic trend across districts was less marked, and only small clusters of infection remained (Fig. 3c–f). As summarised in Supplementary Table 2, the levels of parasite connectivity between the districts in the northern and southern regions did not appear to be fully explained by geographic distance.

P. vivax connectivity in Ethiopia using measures of IBD. Panels (a–f) present cluster plots illustrating the relatedness between infections, as measured using identity by descent (IBD) measures of genetic distance, at thresholds ranging from 0.05 (minimum 5% genome shared) to 0.95 (minimum 95% genome shared, largely reflecting clones). At the 0.05 threshold, all infections aside from SGH-1-357 are connected to at least one other infection. At the 0.05 and 0.075 thresholds, the majority of isolates from Gondar and North Shewa form a distal cluster to the other districts. The isolates from Jimma also display relatively lower connectivity to the other districts. At the 0.1 threshold, geographic trends are less apparent, but multiple connections remain between infections within and between districts, particularly in North Shewa, West Arsi and Sidama. At the 0.25–0.95 thresholds (half-siblings and greater), connections are only observed within districts. The plots are based on data generated using n = 102 independent monoclonal infections.
Neighbour-joining and ADMIXTURE analyses, which use distance measures based on Identity by State (IBS), confirmed subtle patterns of clustering amongst the isolates from Gondar and North Shewa, and Jimma, relative to the other districts (Supplementary Fig. 2a–c). The lowest CV error with ADMIXTURE analysis was at K = 5 (0.034), but there was a limited difference in the absolute number between K = 3 and K = 7 (0.034–0.035) (Supplementary Fig. 2d). At K = 3, aside from Gondar, the majority of isolates in each district exhibited predominant ancestry to the K1 sub-population. In Gondar, all isolates exhibited predominant ancestry to the K2 sub-population. Approximately 29.4% (5/17) of the isolates from North Shewa also exhibited predominant (> 80%) ancestry to K2. The K3 sub-population had the greatest representation in North Shewa, where 29.4% (5/17) of infections had predominant ancestry to this group. A large proportion (17/26, 65.4%) of the infections from Jimma displayed less than 80% ancestry to any given sub-population, rather, showing more mixed ancestry to the K1 and K2 sub-populations.
Heterogeneity in prevalence of variants implicated in chloroquine, antifolate and mefloquine resistance
The prevalence of several variants that have previously been associated with clinical or ex vivo antimalarial drug resistance28, was determined for each of the nine districts with ≥ 4 isolates (Table 2). The most widely characterised candidate is the multidrug resistance 1 (pvmdr1) Y976F variant29,30, a minor modulator of chloroquine (CQ) resistance, had prevalence > 25% in all districts, and > 50% in North Shewa (6/12, 50.0%), West Arsi (24/36, 66.6%) and Gamo (3/6, 50.0%). However, the F1076L variant, which has also been implicated in CQ resistance31 was fixed at 100% frequency in all nine districts. None of the isolates had the pvmdr1 copy number amplification associated with mefloquine resistance. A range of mutations in the dihydrofolate reductase (pvdhfr) and dihydropteroate synthase (pvdhps) genes have been associated with antifolate resistance32,33,34,35. The most common pvdhfr variants observed in Ethiopia were the S58R and S117N mutations, with double mutants present from 50 to 100% of isolates from across the districts. Neither the triple nor the quadruple pvdhfr mutants were observed in any district. At the pvdhps locus, the prevalence of A383G mutations was highly variable between sites, ranging from 0% in Metekel and Sidama to > 67% in East Shewa and Gondar, but no clear geographic trend was observed. No pvdhps A553G mutations were observed in any district.
Substantial difference in frequency of a non-synonymous pvaat1 variant between Ethiopia relative to Thailand and Indonesia
The molecular basis of antimalarial drug resistance is better understood in P. falciparum than P. vivax16. We therefore explored the prevalence of other non-synonymous variants in pvmdr1, pvdhps, pvdhfr and orthologues of several other genes implicated in P. falciparum resistance; pvaat1 (amino acid transporter), pvcrt-o (chloroquine resistance transporter), plasmepsin IV, pvmrp1 and pvmrp2 (multidrug resistance-associated proteins 1 and 2) and pvmdr2 (multidrug resistance protein 2). A summary is provided for Ethiopia and the comparator populations, Thailand and Indonesia, in Supplementary Data 2. Twenty-three non-synonymous variants displayed large differences in frequency, with non-overlapping confidence intervals between Ethiopia and Thailand or Indonesia (Fig. 4). The pvaat1 S25P, three pvdhfr (S117N, S117T, T61M), pvmdr1 S513R, four pvmdr2 variants (A324V, P1466L, V43L and Y514F), pvmrp1 E906Q, two pvmrp2 (E88Q and Y1414H) and one pvdhps (A383G) variant displayed consistently different prevalence (> 20% higher or lower) between Ethiopia and the Asian populations, potentially reflecting Ethiopian-specific adaptations.

Amino acid frequencies in Ethiopia, Thailand and Indonesia at selected P. vivax drug resistance candidates. Each panel presents proportions and corresponding 95% confidence intervals (CIs) for the given amino acid changes. Data are provided on variants that have either (i) previously been associated with drug resistance (association with clinical phenotype) or (ii) are non-synonymous variants in orthologues of P. falciparum drug resistance-associated genes and exhibit substantial differences (non-overlapping CIs) in proportion between Ethiopia and Thailand or Indonesia; the latter class of variants are denoted with (*). For variants that have previously been associated with clinical or ex vivo resistance, frequencies reflect the drug-resistant amino acid, whilst frequencies for the other variants are relative to the reference strain (PvP01).
Extended haplotype homozygosity reveals evidence of selection in the vicinity of pvcrt-o in Ethiopia
Analysis of other gene regions using measures of extended haplotype homozygosity, including the Rsb and iHS measures, provided evidence of recent directional selection. A previous Ethiopian study (n = 17 monoclonal samples) identified a weak signal of selection proximal to pvcrt-o in comparisons against Thailand but not Indonesia25; in the current analysis we focused comparisons of isolates from the same countries using the Rsb metric. Seventeen signals of directional selection were observed in Ethiopia relative to Thailand and four of these were also observed relative to Indonesia (Fig. 5a,b, Supplementary Table 3). Several drug resistance candidates were present within or proximal to these regions of selection, including a signal proximal to pvcrt with extended haplotypes in Ethiopia relative to Thailand (signal), confirming our previous findings (Fig. 5c)25. The signal 1 region includes another putative driver of drug resistance, a prodrug activation and resistance esterase (PvP01_011010). A signal in a putative driver for artemisinin resistance, the pvkelch 10 gene (PvP01_0607800, signal 9), that has previously been detected in Afghanistan, was exhibited in extended haplotypes present in Ethiopia relative to Indonesia36. A third drug-related signal of selection (signal 5) was observed in an orthologue of a new antimalarial target, acyl-CoA-synthetase (PvP01_0409900), with extended haplotypes in Ethiopia relative to Thailand37,38. Other signals of selection encompassed genes with a range of putative functions (detailed in Supplementary Table 3). Amongst these, signals 6 and 14 had peak Rsb scores in immune-related genes. Signal 6, is located in a region including a cluster of serine-repeat antigens (SERA), and merozoite surface protein (MSP) 4 and 5 (PVP01_0418300, PVP01_0418400), which are potential vaccine candidates36,39,40,41,42. The peak at signal 14 is in a gene with unknown function, but the region is downstream from a large MSP7-like gene cluster.

Patterns of extended haplotype homozygosity in Ethiopia. Panels (a) and (b) present Manhattan plots of the Rsb-based cross-population extended haplotype homozygosity (EHH) illustrating regions of divergent selection between Ethiopia and Thailand and Indonesia. Putative signals are numbered. Briefly, putative drivers include; chloroquine resistance transporter (pvcrt, PvP01_0109300) and prodrug activation and resistance esterase (PvP01_011010) at signal 1, lysine specific histone demethylase (PVP01_0118300) at signal 2, peptide chain release factor 2 (PvP01_0309000) at signal 3, phosphoinositide-binding protein PX1 (PvP01_0316400) at signal 4, acyl-CoA-synthetase (PvP01_0409900) at signal 5, merozoite surface protein (MSP) 4 and 5 (PVP01_0418300, PVP01_0418400) at signal 6, zinc finger protein (PvP01_0517200) at signal 7, serine/threonine protein phosphatase (PvP01_0603400) at signal 8, pvkelch protein K10 (PvP01_0607800) at signal 9, a tRNA (PVP01_0711200) at signal 10, PvP01_0824800 and PvP01_1115800 (unknown functions) at signals 11 and 12, oligomeric golgi complex subunit 4 (PvP01_1133300) at signal 13, an MSP7-like gene cluster at signal 14, intergenic region at signal 15, actin related protein ARP4 (PvP01_1326200) at signal 16 and liver-specific protein (PvP01_1330800) at signal 17. Panel (c) illustrates the comparative decay in EHH in the pvcrt-o region of signal 1. The dashed vertical grey line indicates the position at which the peak Rsb score was observed. The pink shaded region denotes the pvcrt-o region. While the peak Rsb is not located within pvcrt-o, haplotype homozygosity appears to decay less rapidly upstream (i.e., in the direction of pvcrt-o) than downstream of the signal peak. Ethiopia retains moderate haplotype homozygosity (EHHS > 0.1) within the pvcrt-o region. Panel (d) presents a Manhattan plot of the integrated haplotype score (iHS) illustrating regions under recent directional selection in Ethiopia alone. Only a single peak was detected in an intergenic region at signal 18. The data was derived from analyses on independent samples from Ethiopia (n = 102), Indonesia (n = 110) and Thailand (n = 87).
The integrated haplotype score (iHS) was also used to identify regions under apparent directional selection in Ethiopia as evidenced by relatively extended haplotypes flanking the alternate alleles at a given SNP. Only one major signal of directional selection (signal 18) was observed in Ethiopia, in a 3.5 kb region on chromosome 14 comprising 2 genes (Fig. 5d, Supplementary Table 3). The peak signal was in an intergenic region between two genes encoding Plasmodium exported proteins, PvP01_1470400 and PvP01_1470500.
High frequency of duffy binding protein 1 copy number amplification
The most prevalent copy number (CN) variant observed in Ethiopia was present in the P. vivax duffy binding protein 1 (pvdbp1), detected in 67.8% (93/112) of the analysable samples. Most pvdbp1 CN amplifications harboured the previously described Cambodian breakpoint (88.2%, 82/93), with the remaining samples (11.8%, 11/93) having the Malagasy breakpoint (Supplementary Data 3)43. The number of pvdbp1 copies in the amplifications ranged from 2 to 5 copies. Although some variation was observed between districts, all had a prevalence of pvdbp1 CN amplification > 60% (Fig. 6a) with no apparent geographic trends.

Prevalence and diversity of duffy binding protein 1 copy number amplifications in Ethiopia. Panel (a) illustrates the prevalence of the pvdbp1 copy number amplification by district. The error bars reflect Clopper-Pearson confidence intervals. A high prevalence of pvdbp1 copy number amplification (> 60%) was observed in all districts assessed. However, as illustrated by the standard error bars, sample size was constrained in districts such as Metekel and East Shewa. Panel (b) presents a heatmap illustrating the divergence between pvdbp1 copies in monoclonal infections with copy number amplifications. Rows indicate samples and columns indicate amino acid changes conferred by underlying SNPs. Genotypes are shown as reference allele frequencies on a scale of 0 in red (homozygote alternative allele) to 1 in blue (homozygous reference allele). As the infections are monoclonal, heterozygotes (trending towards orange) are indicative of sequence divergence between the pvdbp1 copies. Infections are clustered by genetic relatedness in the given sequence region. Sample identifiers (row labels) are colour-coded by district (as in panel a), and the number of pvdbp1 copies is indicated in parentheses. Majority of samples have few heterozygotes (mostly red or blue genotypes) but several samples deriving from a range of districts have extensive divergence between pvdbp1 copies (many orange genotypes). Panel (a) was generated using data on 102 independent samples, and panel (b) was generated using data on 102 independent, monoclonal samples with pvdbp1 amplification.
The monoclonal isolates with pvdbp1 CN amplification (n = 67) were further investigated to evaluate the additional genetic variation created by the extra gene copies. Amongst the 67 monoclonal samples, a mean of 3.4 heterozygous SNPs was observed in the CN region, indicative of modest sequence differences between the copies (Fig. 6b). However, the range of heterozygous SNPs ranged up to a maximum of 23 SNPs, highlighting the potential for extensive diversity between the pvdbp1 copies.
Discussion
Our large genomic study includes geographically widespread P. vivax Ethiopian isolates. It reveals high diversity in all geographic regions assessed, complex patterns of parasite connectivity between districts, and strong evidence of selection in a region proximal to the pvcrt-o locus. We confirm the high prevalence of the pvdbp1 CN amplification in all geographic regions assessed. Herein we describe the epidemiological processes that may be shaping the observed population diversity and structure, and present hypotheses for the observed signals of selection.
Ethiopia has reported a steady rise of P. vivax infections from 2019 to 2021, and reported data systematically underestimates the true burden of infection as it does not account for the hidden reservoir of asymptomatic infections1,3. Current approaches to monitor P. vivax epidemiology using methods such as microscopy and rapid diagnostic tests (RDTs) are limited in detecting sub-microscopic and asymptomatic infections44. The dormant liver stage (hypnozoites) and splenic reservoirs further complicate efforts to infer the true toll of P. vivax infection in Ethiopia. Population genetic metrics can provide complimentary insights into parasite transmission, with P. falciparum studies revealing a positive correlation between transmission intensity and prevalence of polyclonal infections45. The situation in P. vivax is more complex, attributable in part to the hypnozoite reservoir17,21. In our study, 26% of infections were polyclonal, similar to our previous Ethiopian estimate of ~ 30% polyclonal infections25. These frequencies are lower than that observed in areas of intense P. vivax transmission such as Papua Indonesia (48%), but higher than in pre-elimination settings such as Malaysia (16%) or Panama (0%)21,24,45. A population genomic study conducted in northern Ethiopia (across Amhara, Gambella and Tigray) in 2017–18 detected a slightly lower level of polyclonal P. falciparum infections (~ 18%) potentially reflecting a contribution of the liver-stage reservoir to P. vivax transmission, but this needs to be investigated in co-endemic populations and with consideration of seasonal changes46. Our data also revealed marked polyclonal heterogeneity at the district level (0–55%), consistent with Ethiopia’s marked variation in malaria endemicity25. Although the sample size was limited in several districts, in the four districts with ≥ 10 isolates, there was a positive correlation between the API-based malaria endemicity (defined by the Ethiopian National Malaria Elimination Program (NMEP) and the frequency of polyclonal infections. In these four districts, polyclonality ranged from 12% in North Shewa (a pre-elimination settings), to 55% in Gondar (~ 400 km from North Shewa), more comparable to the intense transmission in Papua. Our findings concur with a microsatellite-based genotyping study, which revealed heterogeneity in P. vivax infection diversity between relatively proximal districts in southern Ethiopia17. Ecological, climatic and demographic factors are likely to impact malaria transmission in this region, but the limited sample size in several districts constrained our ability to conduct in-depth investigation of these factors. Further studies are needed with high-throughput genotyping across dense sample sets.
Our analysis of the relatedness (IBD) between the clones within polyclonal infections enabled further insights into P. vivax transmission, specifically concerning co-transmission (single mosquito inoculation carrying mixtures of parasite genomes) and superinfection (multiple mosquito inoculations)47. In blood meals with mixtures of parasite genomes, the obligate meiotic stage generates recombinant sporozoites that share parents, hence clones with evidence of recent IBD are more likely to have derived from the same mosquito inoculation than from different inoculations. Owing to the liver-stage reservoir, mixtures of unrelated P. vivax genomes can arise from infectious bites in close succession, or a recent inoculum combined with a reactivated hypnozoite from an older bite; both cases involve multiple inoculums. In concordance with our previous study in Ethiopia, almost half of all polyclonal infections carried at least two highly related clones (siblings or half-siblings, sharing ≥ 25% genomic IDB), suggestive of co-transmission events, with the remaining isolates presumably reflecting superinfections25. These findings infer a high proportion of both co-infection and superinfection events in Ethiopia. There was broad heterogeneity in the prevalence of putative superinfections between districts (0–100%). However, sample size was modest in several sites and our definition of superinfection (within-host IBD < 25%) may be imperfect and needs further exploration. Comparative evaluation of the IBD patterns in polyclonal infections in co-endemic P. vivax and P. falciparum populations will provide useful insights into the contribution of untreated hypnozoites to rates of P. vivax co-infection and superinfection, informing on transmission reduction priorities. It will also be important to explore within-host IBD patterns in sub-patent and asymptomatic infections, which may exhibit different dynamics than the symptomatic, patent reservoir48.
Information on the key drivers and barriers of infection spread between communities is critical for the national malaria elimination plan to decide how and where to prioritise interventions for maximum transmission reduction. The wide heterogeneity in the composition and abundance of Anopheline vector species, landscape features, agricultural practices and ethnic/cultural distributions complicate prediction of the main reservoirs and routes of parasite spread within and between communities of Ethiopia. Hence parasite genetic data has great potential to provide information on parasite connectivity between communities49,50,51. Previous studies using microsatellite data to measure IBS between P. vivax infections from different communities observed that geographic distance was not a major determinant of P. vivax infection spread in Ethiopia17,52. IBD has greater potential than IBS to capture connectivity between parasites in highly recombining species such as Plasmodium spp.53,54. Using both IBS and IBD measures on our genomic dataset, we confirmed that geographic distance was not a major driver of P. vivax connectivity in Ethiopia. Whilst isolates from the northern districts of Gondar and Shewa Robit could be differentiated from the southern districts, the northern district of Metekel had limited connectivity with neighbouring Gondar, instead exhibiting greater connectivity with the southern districts. This area is the epicentre of the Grand Ethiopian Renaissance Dam (GERD), the biggest hydroelectric dam in Africa, which has been under construction since 2011, employing between 8500–12,000 people from across Ethiopia at its peak. The high connectivity between the P. vivax populations in Metekel and the southern districts may therefore reflect a large influx of migrant workers from these regions. Although Gondar and Shewa Robit are > 400 km apart, the high connectivity is consistent with historical cultural ties between these districts55. Another distinct pattern observed in our dataset was moderate differentiation between Jimma and neighbouring districts in the south of the country. The patterns of gene flow with Jimma may reflect dense forest in the region that could impede movement of people from nearby districts. Our results suggest that economic and cultural factors are likely to be important drivers of human mobility and associated P. vivax infection spread in Ethiopia. These factors can be difficult to predict but combining parasite genetics with other data on human mobility such as mobile phone data, may provide critical information on connectivity between communities, as has been demonstrated in P. falciparum studies49,50.
Defining antimalarial drug resistance in P. vivax is challenging16,28. Clinical efficacy of treatment regimens for P. vivax is confounded by recurrent infections which can arise from recrudescence, reinfection or relapse. Chloroquine (CQ) remains the mainstay of treatment for P. vivax in Ethiopia and most vivax endemic countries, although high grade CQ resistance (CQR) has been reported from Indonesia, Papua New Guinea and Malaysia. In most other endemic areas, there have also been sporadic reports of low grade CQR56. Although some therapeutic efficacy studies have documented declining CQ efficacy in Ethiopia7,8,9,10,11,13,14,57, others report sustained high efficacy12,58,59,60. The local P. vivax population has also been subject to selection pressure from artemether-lumefantrine and other antimalarials targeting the co-endemic P. falciparum population. Sulfadoxine-pyrimethamine (SP) and CQ were withdrawn from treatment guidelines for P. falciparum more than three decades ago owing to their poor efficacy61. However, SP is still recommended for intermittent preventive treatment in pregnancy (IPTp), infancy (IPTi) and childhood (IPTc) for all malaria species62,63,64. Our study revealed a high frequency of pvdhfr double mutants (58R/117N) in all districts evaluated (50–100%). Studies have shown that these pvdhfr double mutants can induce up to 460-fold increase in resistance to pyrimethamine65. In contrast to many Southeast Asian populations, no isolates with triple or quadruple pvdhfr mutations were observed in Ethiopia, suggesting lower selective pressure from SP25. The apparent lower selective pressure in Ethiopia may reflect a lower rate of drug implementation than in Thailand or Indonesia. At the pvdhps locus, the wildtype 553A variant predominated, but heterogeneity was observed between districts in the prevalence of the resistance-associated 383G variant. Further evaluation of the impact of the combined 58R/117N/553A/383G genotype on SP efficacy in IPTp and IPTi is required.
Our study also determined the prevalence of P. vivax multidrug resistance 1 (pvmdr1) polymorphisms. In keeping with the limited use of mefloquine in Ethiopia, there was no evidence of pvmdr1 copy number amplification, which has been associated with mefloquine resistance19. Single nucleotide polymorphisms at pvmdr1 976 and 1076 have been implicated in CQR29,31. There was a high prevalence of the pvmdr1 976F mutation (25–67%), and pvmdr1 1076L was at fixation6,16,29. However, the significance of these mutations is uncertain as they may be only minor modulators of CQR. The major molecular determinant of CQR in P. falciparum is the chloroquine resistance transporter (pfcrt)66,67. Although several studies have proposed that the pfcrt orthologue (pvcrt-o) has a role in P. vivax CQR, this remains contentious6,16. In a previous genomic analyses, we reported extended haplotype homozygosity in a region proximal to pvcrt-o in Ethiopia relative to Thailand (where CQR prevalence was low) but not Papua Indonesia (where CQR prevalence was high); however, the study was constrained by small sample size with only 17 monoclonal Ethiopian isolates25. In the current study, we confirmed the signal in the pvcrt-o region using 102 monoclonal Ethiopian isolates. However, this signal region contains other gene candidates, including a prodrug activation and resistance esterase (PvP01_011010), which may also be potential drivers of the observed haplotype homozygosity. The epicentre of CQR in P. vivax is in Papua, Indonesia, and yet comparisons between Indonesia and Thailand found no evidence of extended haplotypes in the pvcrt-o region24,25. This difference could reflect variation in demographic or selective pressures, or potentially different mechanisms of CQR between populations. Further functional studies of pvcrt-o are needed to explore this further68.
A recent study has implicated the amino acid transporter 1 gene (pfaat1) in the evolution of chloroquine resistance in P. falciparum27. Gene editing demonstrates that pfaat1 S258L potentiates CQ resistance at a fitness cost, while a common southeast Asian variant (pfaat1 F313S) reduces CQ resistance while restoring fitness27. We found no evidence of extended haplotypes at pvaat1 in Ethiopia (or Thailand or Indonesia). However, we did observe substantially higher frequency of a non-synonymous variant (pvaat1 S25P) in Ethiopia (96%) than Thailand (35%) or Indonesia (53%), potentially reflecting local differences in CQ resistance evolution. Three-dimensional protein structure models of pvaat1 will be helpful to discern the impact of pvaat1 S25P on drug transportation in P. vivax.
Our study also highlighted selection of regions coding for other putative drivers of drug resistance. Most prominent of these was the P. vivax kelch 10 gene (PvP01_0607800), an orthologue of P. falciparum kelch13, which has been associated with artemisinin resistance69,70. However, the implications of this with regard for artemisinin resistance in P. vivax are unclear; clinical efficacy studies have shown that artemisinin-based combination therapies (ACTs) including artemether-lumefantrine (AL), retain potent efficacy against P. falciparum and P. vivax in Ethiopia11. A recent study revealed 8% prevalence of a P. falciparum candidate artemisinin partial resistance kelch13 R622I mutation across three regions of Ethiopia46. Although the R622I mutant has not been validated as a determinant of artemisinin resistance, close monitoring of both P. falciparum and co-endemic P. vivax populations for early assigns of ACT resistance is a priority.
The low prevalence of P. vivax in most of sub-Saharan Africa has been attributed to the predominance of the human Duffy negative genotype, which prevents expression of the Duffy Antigen Receptor for Chemokine (DARC) in red blood cells (RBCs)2,71,72. For several decades, DARC was thought to be critical for P. vivax invasion of human RBCs, mediated by a parasite duffy binding protein encoded by pvdbp171. However, there are increasing reports of P. vivax infection in DARC (Duffy) negative individuals in Ethiopia73. The rate of Duffy negativity ranges from 3 to 35% in Ethiopia, providing a setting in which Duffy heterozygous individuals could prime P. vivax adaptations enabling invasion of Duffy negative RBCs52,73,74,75. An alternative hypothesis is that other pathways may facilitate P. vivax to invade Duffy negative individuals76. There was a high prevalence of pvdbp1 copy number amplifications (67.8% infections carrying two or more copies) in all districts assessed, with evidence of at least two independent origins, with some isolates exhibiting up to 5 gene copies; these findings suggest major adaptive functions across Ethiopia. It’s possible that pvdbp1 amplification may enable low-affinity binding of P. vivax to Duffy negative RBCs via an alternative receptor to DARC26, or pvdbp1 amplification may allow for mutations in the extra gene copies, which could enhance diversity and immune evasion26. Indeed, we observed evidence of moderate diversity between pvdbp1 copies in several infections evaluated. In line with the immune evasion hypothesis, a study in Cambodia found evidence that pvdbp1 amplification reducing humoral immunity against P. vivax72. The prevalence of pvdbp1 amplification in Ethiopia is the highest reported globally77, and may reflect an adaptation supporting both RBC invasion and immune evasion mechanisms.
In summary, the genomic architecture of P. vivax in Ethiopia highlights adaptations of potential public health concern in an endemic setting with evidence of stable transmission and long-distance spread of infection. Our findings highlight the need for more dense and geographically widespread molecular data that can be generated in a timely manner to facilitate effective surveillance and response.
Methods
Samples and study sites
All genomic data used in the study was derived from the open-access malaria Genomic Epidemiology Network (MalariaGEN) P. vivax genome variation project (Pv4.0) dataset77. The Ethiopian P. vivax genomic data were derived from patient isolates collected in the framework of clinical surveys and cross-sectional studies conducted in nine districts across northern and southwestern Ethiopia between 2012 and 2016 (Fig. 1). Briefly, published P. vivax genomes were obtained from cross-sectional surveys undertaken in Arbaminch, Badowacho, Hawassa and Halaba between May and November 2013, and Jimma between September and November 201618,25. Additional samples were collected from two frameworks: a previously described clinical survey of Tafenoquine efficacy in P. vivax-infected patients recruited from Gondar and Jimma in 201678, and a cross-sectional survey conducted in West Arsi, East Shewa (Adama) and Shewa Robit (North Shewa) between February 2013 and October 201577. Details on the publication status of the samples can be found in Supplementary Data 1. Based on diverse ecological risk factors that correspond to a range of transmission intensity and pattern, malaria transmission was stratified into five strata: high malaria endemicity, moderate malaria endemicity, low malaria endemicity, very low malaria endemicity and malaria-free areas79. Three of our study sites, Sidama, Gamo and Metekel represent areas of highly endemic malaria, with stable year-round transmission, supported by hot, humid, tropical climates. Hadiya, Jimma and West Arsi represent moderate malaria endemic areas with moderate, seasonal transmission. Shewa Robit, Gondar and East Shewa (Adama) are areas with overall very low malaria endemicity, but a short period of intense malaria transmission. In Ethiopia, the first line antimalarial for treating P. vivax infection is chloroquine whereas artemether-lumefantrine is used to treat monoclonal P. falciparum or mixed-species (P. falciparum, P. vivax) infections.
Comparative analyses were undertaken using MalariaGEN Pv4.0 data from Thailand and Papua Indonesia25. The Thai population represented a region with low-grade chloroquine resistance, while the Papua Indonesia population represented a region with high-grade chloroquine resistance80,81. The samples were collected from symptomatic patients attending outpatient clinics in Tak Province, Thailand (2006–2013) and Mimika district, Papua Indonesia (2011–2014). The frontline treatment for P. vivax infection at the time of the enrollments was chloroquine plus primaquine in Thailand and dihydroartemisinin-piperaquine plus primaquine in Indonesia.
Isolates and sequence data used for analysis.
The study used genome-wide Single Nucleotide Polymorphism (SNP) and Copy Number Variant (CNV) data from the MalariaGEN Pv4.0 release77. All Pv4.0 data are derived from P. vivax patient samples that were sequenced using Illumina platforms with paired end reads. In brief, after exclusion of any human reads, P. vivax sequence data from MalariaGEN partners and selected published data sets were mapped to the P. vivax P01 v1 reference genome using bwa82,83. GATK Best Practices Workflows were used to call SNPs and minor insertions and deletions (indels)84. The resulting Variant Calling Format (VCF) file, describing ~ 4.5 million variants in 1895 P. vivax samples is publicly available on the MalariaGEN website (https://www.malariagen.net/data/open-dataset-plasmodium-vivax-v4.0). CNVs due to large tandem duplications (> 3 kbp) were genotyped using a two-stage process where breakpoints were first discovered at base pair resolution using a combination of read depth and split reads and then sample were genotyped at these discovered breakpoints using a combination of read depth and read pairs mapped in a tail-to-tail configuration. Details on these CNVs, including copy number amplifications of the pvmdr1 and pvdbp1 genes are also available within the open access Pv4.0 data release77.
From the initial Pv4.0 VCF, only independent (i.e., no recurrence pairs or duplicates) samples from Ethiopia (n = 159), Thailand (n = 129) and Indonesia (n = 191) were extracted. Sample and SNP filtering processes were performed to exclude low-quality variants and reduce genotyping failures (defined as positions with 0) biallelic SNPs with < 5% sample failure rate and samples with < 15% genotype failure at the given SNPs. The final population genomic analysis set (from Ethiopia (n = 137), Thailand (n = 104), and Indonesia (n = 111)) contained 448 samples and 410,900 SNPs. The list of Ethiopian isolates along with European Nucleotide Archive accession numbers is provided in Supplementary Table 1. For candidate drug resistance analysis, a second VCF was generated that contained all high quality (VQSLOD > = 0) variants (with no filtering by genotyping failure or biallelic status) within previously described drug resistance candidates, with no further sample filtering28.
Data analysis
The within-sample F statistic (Fws) was used to evaluate within-host infection complexity85,86. Fws > 0.95 was utilised as a cutoff to represent monoclonal infections. Within-host infection complexity was further explored using DEploid software to deconvolve polyclonal infections87,88. Briefly, all Ethiopian samples were used to create a population level allele frequency (PLAF) file. The samples were then scanned with DEploid to identify samples with more than one clone with 10% or more proportions. To create a reference panel, only samples with Fws > 0.95 and only one clone with 10% or more proportions were included. Samples that were not included in the reference panel were deconvolved with DEploid-BEST (https://github.com/DEploid-dev/DEploid). Deconvolved haplotypes were only derived on clones making up a minimum 10% of the total infection composition as estimated by allele frequency. Using the deconvolved haplotype reconstructions, hmmIBD software was used to determine the identity by descent (IBD) between the clones within each infection using default parameters89. The genome-wide patterns of within-host diversity were also illustrated with Manhattan plots of the non-reference allele frequency (NRAF). All further analyses were restricted to the monoclonal isolates (Fws > 0.95).
Spatial patterns of infection relatedness within Ethiopia were assessed using IBD measures between infections, calculated using hmmIBD89. IBD thresholds ranging from 0.05 (minimum 5% genome shared) to 0.95 (minimum 95% genome shared) were illustrated using network plots created using the R-based igraph package (https://igraph.org). Parasite relatedness was also assessed with neighbour-joining (NJ) analysis, using the R Adegenet package to compute a pairwise distance matrix and iTOL software to plot a tree90. The distance matrix was also used to conduct principal coordinate analysis (PCoA) using the R-based adegenet package91. ADMIXTURE analysis was used to infer population structure, and the cross-validation error was used to determine the most likely number of subpopulations (K)92.
Signals of selection were explored using the R-based rehh software to calculate the integrated haplotype score (iHS) and the Rsb measure of cross-population extended haplotype homozygosity (XP-EHH). The Thai and Indonesian populations were used as comparator populations for the XP-EHH analysis. Using similar thresholds to previous studies, signals supported by ≥ 3 SNPs above a threshold –log10 (P value) > 4 within 50 kb of one another and with an overall SNP density < 10 kb per SNP are numbered25,36.
Ethics
As detailed in the MalariaGEN Pv4.0 data release, all P. vivax patient isolates used in the study were collected with local ethics approval, and written informed consent was obtained from all participants or a legal guardian where participants were ≤ 18 years of age77. In addition to the Pv4.0 release, several of the Ethiopian genomes have been published elsewhere25,78,93. Ethical approval for patient sampling from the new study sites in East Shewa, West Arsi and Shewarobit was granted by the ethics boards of Aklilu Lemma Institute of Pathobiology, Addis Ababa University, Armauer Hansen Research Institute, the national research ethics, the London School of Hygiene and Tropical Medicine. All methods were performed in accordance with the guidelines and regulations of the listed ethics committees.
Data availability
The raw sequencing data for the 137 high-quality Ethiopian P. vivax samples described in the study are all openly accessible in the European Nucleotide Archive, with accession codes detailed in Supplementary Data 1. As described in the methods, the P. vivax genotyping data used for the Ethiopian and global analyses all derives from the MalariaGEN Pv4.0 dataset and is accessible as a Variant Calling Format (VCF) file (describing ~ 4.5 million variants in 1895 worldwide P. vivax genomes) on the MalariaGEN website at https://www.malariagen.net/data/open-dataset-plasmodium-vivax-v4.0.
References
-
World Health Organization. World Malaria Report 2022 (World Health Organization, 2022).
-
Twohig, K. A. et al. Growing evidence of Plasmodium vivax across malaria-endemic Africa. PLoS Negl. Trop. Dis. 13, e0007140. https://doi.org/10.1371/journal.pntd.0007140 (2019).
Google Scholar
-
Tadesse, F. G. et al. The relative contribution of symptomatic and asymptomatic Plasmodium vivax and Plasmodium falciparum infections to the infectious reservoir in a low-endemic setting in Ethiopia. Clin. Infect. Dis. 66, 1883–1891 (2018).
Google Scholar
-
Keffale, M. et al. Serological evidence for a decline in malaria transmission following major scale-up of control efforts in a setting selected for Plasmodium vivax and Plasmodium falciparum malaria elimination in Babile district, Oromia, Ethiopia. Trans. R. Soc. Trop. Med. Hyg. 113, 305–311 (2019).
Google Scholar
-
Price, R. N., Commons, R. J., Battle, K. E., Thriemer, K. & Mendis, K. Plasmodium vivax in the era of the shrinking P. falciparum map. Trends Parasitol. 36, 560–570 (2020).
Google Scholar
-
Auburn, S., Cheng, Q., Marfurt, J. & Price, R. N. The changing epidemiology of Plasmodium vivax: Insights from conventional and novel surveillance tools. PLoS Med. 18, e1003560. https://doi.org/10.1371/journal.pmed.1003560 (2021).
Google Scholar
-
Teka, H. et al. Chloroquine-resistant Plasmodium vivax malaria in Debre Zeit, Ethiopia. Malaria J. 7, 1–8 (2008).
Google Scholar
-
Assefa, M., Eshetu, T. & Biruksew, A. Therapeutic efficacy of chloroquine for the treatment of Plasmodium vivax malaria among outpatients at Hossana Health Care Centre, southern Ethiopia. Malar. J. 14, 1–8 (2015).
Google Scholar
-
Schunk, M. et al. High prevalence of drug-resistance mutations in Plasmodium falciparum and Plasmodium vivax in southern Ethiopia. Malar. J. 5, 54. https://doi.org/10.1186/1475-2875-5-54 (2006).
Google Scholar
-
Golassa, L., Erko, B., Baliraine, F. N., Aseffa, A. & Swedberg, G. Polymorphisms in chloroquine resistance-associated genes in Plasmodium vivax in Ethiopia. Malar. J. 14, 1–5 (2015).
Google Scholar
-
Abreha, T. et al. Comparison of artemether-lumefantrine and chloroquine with and without primaquine for the treatment of Plasmodium vivax infection in Ethiopia: A randomized controlled trial. PLoS Med. 14, e1002299 (2017).
Google Scholar
-
Shumbej, T., Jemal, A., Worku, A., Bekele, F. & Weldesenbet, H. Therapeutic efficacy of chloroquine for treatment of Plasmodium vivax malaria cases in Guragae zone southern Central Ethiopia. BMC Infect. Dis. 19, 413. https://doi.org/10.1186/s12879-019-4055-7 (2019).
Google Scholar
-
Tulu, A. N., Webber, R. H., Schellenberg, J. A. & Bradley, D. J. Failure of chloroquine treatment for malaria in the highlands of Ethiopia. Trans. R. Soc. Trop. Med. Hyg. 90, 556–557 (1996).
Google Scholar
-
Mekonnen, S. K. et al. Return of chloroquine-sensitive Plasmodium falciparum parasites and emergence of chloroquine-resistant Plasmodium vivax in Ethiopia. Malar. J. 13, 1–9 (2014).
Google Scholar
-
Ketema, T., Bacha, K., Getahun, K. & Bassat, Q. In vivo efficacy of anti-malarial drugs against clinical Plasmodium vivax malaria in Ethiopia: A systematic review and meta-analysis. Malar. J. 20, 1–19 (2021).
Google Scholar
-
Buyon, L. E., Elsworth, B. & Duraisingh, M. T. The molecular basis of antimalarial drug resistance in Plasmodium vivax. Int. J. Parasitol. Drugs Drug Resist. 16, 23–37. https://doi.org/10.1016/j.ijpddr.2021.04.002 (2021).
Google Scholar
-
Getachew, S. et al. Variation in complexity of infection and transmission stability between neighbouring populations of Plasmodium vivax in Southern Ethiopia. PLoS One 10, e0140780 (2015).
Google Scholar
-
Ford, A. et al. Whole genome sequencing of Plasmodium vivax isolates reveals frequent sequence and structural polymorphisms in erythrocyte binding genes. PLoS Negl. Trop. Dis. 14, e0008234 (2020).
Google Scholar
-
Auburn, S. et al. Genomic analysis reveals a common breakpoint in amplifications of the Plasmodium vivax multidrug resistance 1 locus in Thailand. J. Infect. Dis. 214, 1235–1242. https://doi.org/10.1093/infdis/jiw323 (2016).
Google Scholar
-
Winter, D. J. et al. Whole genome sequencing of field isolates reveals extensive genetic diversity in Plasmodium vivax from Colombia. PLoS Negl. Trop. Dis. 9, e0004252. https://doi.org/10.1371/journal.pntd.0004252 (2015).
Google Scholar
-
Auburn, S. et al. Genomic analysis of a pre-elimination Malaysian Plasmodium vivax population reveals selective pressures and changing transmission dynamics. Nat. Commun. 9, 2585 (2018).
Google Scholar
-
Parobek, C. M. et al. Selective sweep suggests transcriptional regulation may underlie Plasmodium vivax resilience to malaria control measures in Cambodia. Proc. Natl. Acad. Sci. 113, E8096–E8105 (2016).
Google Scholar
-
Hupalo, D. N. et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat. Genet. 48, 953–958 (2016).
Google Scholar
-
Pearson, R. D. et al. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet. 48, 959–964 (2016).
Google Scholar
-
Auburn, S. et al. Genomic analysis of Plasmodium vivax in southern Ethiopia reveals selective pressures in multiple parasite mechanisms. J. Infect. Dis. 220, 1738–1749 (2019).
Google Scholar
-
Gunalan, K. et al. Role of Plasmodium vivax duffy-binding protein 1 in invasion of duffy-null Africans. Proc. Natl. Acad. Sci. U.S.A. 113, 6271–6276. https://doi.org/10.1073/pnas.1606113113 (2016).
Google Scholar
-
Amambua-Ngwa, A. et al. Chloroquine resistance evolution in Plasmodium falciparum is mediated by the putative amino acid transporter AAT1. Nat. Microbiol. https://doi.org/10.1038/s41564-023-01377-z (2023).
Google Scholar
-
Price, R. N., Auburn, S., Marfurt, J. & Cheng, Q. Phenotypic and genotypic characterisation of drug-resistant Plasmodium vivax. Trends Parasitol. 28, 522–529. https://doi.org/10.1016/j.pt.2012.08.005 (2012).
Google Scholar
-
Suwanarusk, R. et al. Amplification of pvmdr1 associated with multidrug-resistant Plasmodium vivax. J. Infect. Dis. 198, 1558–1564 (2008).
Google Scholar
-
Suwanarusk, R. et al. Chloroquine resistant Plasmodium vivax: In vitro characterisation and association with molecular polymorphisms. PLoS One 2, e1089 (2007).
Google Scholar
-
Rungsihirunrat, K., Muhamad, P., Chaijaroenkul, W., Kuesap, J. & Na-Bangchang, K. Plasmodium vivax drug resistance genes; Pvmdr1 and Pvcrt-o polymorphisms in relation to chloroquine sensitivity from a malaria endemic area of Thailand. Korean J. Parasitol. 53, 43–49. https://doi.org/10.3347/kjp.2015.53.1.43 (2015).
Google Scholar
-
Marfurt, J. et al. Molecular markers of in vivo Plasmodium vivax resistance to amodiaquine plus sulfadoxine-pyrimethamine: Mutations in pvdhfr and pvmdr1. J. Infect. Dis. 198, 409–417 (2008).
Google Scholar
-
Auliff, A. et al. Amino acid mutations in Plasmodium vivax DHFR and DHPS from several geographical regions and susceptibility to antifolate drugs. Am. J. Trop. Med. Hyg. 75, 617–621 (2006).
Google Scholar
-
Korsinczky, M. et al. Sulfadoxine resistance in Plasmodium vivax is associated with a specific amino acid in dihydropteroate synthase at the putative sulfadoxine-binding site. Antimicrob. Agents Chemother. 48, 2214–2222 (2004).
Google Scholar
-
Imwong, M. et al. Limited polymorphism in the dihydropteroate synthetase gene (dhps) of Plasmodium vivax isolates from Thailand. Antimicrob. Agents Chemother. 49, 4393–4395 (2005).
Google Scholar
-
Benavente, E. D. et al. Distinctive genetic structure and selection patterns in Plasmodium vivax from South Asia and East Africa. Nat. Commun. 12, 3160. https://doi.org/10.1038/s41467-021-23422-3 (2021).
Google Scholar
-
Summers, R. L. et al. Chemogenomics identifies acetyl-coenzyme A synthetase as a target for malaria treatment and prevention. Cell Chem. Biol. 29, 191-201 e198. https://doi.org/10.1016/j.chembiol.2021.07.010 (2022).
Google Scholar
-
Prata, I. O. et al. Plasmodium falciparum acetyl-CoA synthetase is essential for parasite intraerythrocytic development and chromatin modification. ACS Infect. Dis. 7, 3224–3240. https://doi.org/10.1021/acsinfecdis.1c00414 (2021).
Google Scholar
-
Black, C. G., Wang, L., Hibbs, A. R., Werner, E. & Coppel, R. L. Identification of the Plasmodium chabaudi homologue of merozoite surface proteins 4 and 5 of Plasmodium falciparum. Infect. Immun. 67, 2075–2081 (1999).
Google Scholar
-
Black, C. G., Wang, L., Wu, T. & Coppel, R. L. Apical location of a novel EGF-like domain-containing protein of Plasmodium falciparum. Mol. Biochem. Parasitol. 127, 59–68 (2003).
Google Scholar
-
Woodberry, T. et al. Antibodies to Plasmodium falciparum and Plasmodium vivax merozoite surface protein 5 in Indonesia: Species-specific and cross-reactive responses. J. Infect. Dis. 198, 134–142 (2008).
Google Scholar
-
Perraut, R. et al. Association of antibody responses to the conserved Plasmodium falciparum merozoite surface protein 5 with protection against clinical malaria. PLoS One 9, e101737 (2014).
Google Scholar
-
Hostetler, J. B. et al. Independent origin and global distribution of distinct Plasmodium vivax Duffy binding protein gene duplications. PLoS Negl. Trop. Dis. 10, e0005091 (2016).
Google Scholar
-
Tadesse, F. G. et al. Submicroscopic carriage of Plasmodium falciparum and Plasmodium vivax in a low endemic area in Ethiopia where no parasitaemia was detected by microscopy or rapid diagnostic test. Malar. J. 14, 303. https://doi.org/10.1186/s12936-015-0821-1 (2015).
Google Scholar
-
Buyon, L. E. et al. Population genomics of Plasmodium vivax in Panama to assess the risk of case importation on malaria elimination. PLoS Negl. Trop. Dis. 14, e0008962. https://doi.org/10.1371/journal.pntd.0008962 (2020).
Google Scholar
-
Fola, A. A. et al. Plasmodium falciparum resistant to artemisinin and diagnostics have emerged in Ethiopia. Nat. Microbiol. 8, 1911–1919. https://doi.org/10.1038/s41564-023-01461-4 (2023).
Google Scholar
-
Bright, A. T. et al. A high resolution case study of a patient with recurrent Plasmodium vivax infections shows that relapses were caused by meiotic siblings. PLoS Negl. Trop. Dis. 8, e2882. https://doi.org/10.1371/journal.pntd.0002882 (2014).
Google Scholar
-
Pava, Z. et al. Passively versus actively detected malaria: Similar genetic diversity but different complexity of infection. Am. J. Trop. Med. Hyg. 97, 1788–1796. https://doi.org/10.4269/ajtmh.17-0364 (2017).
Google Scholar
-
Tessema, S. et al. Using parasite genetic and human mobility data to infer local and cross-border malaria connectivity in Southern Africa. Elife https://doi.org/10.7554/eLife.43510 (2019).
Google Scholar
-
Chang, H. H. et al. Mapping imported malaria in Bangladesh using parasite genetic and human mobility data. Elife https://doi.org/10.7554/eLife.43481 (2019).
Google Scholar
-
Noviyanti, R. et al. Implementing parasite genotyping into national surveillance frameworks: Feedback from control programmes and researchers in the Asia-Pacific region. Malar. J. 19, 271. https://doi.org/10.1186/s12936-020-03330-5 (2020).
Google Scholar
-
Lo, E. et al. Transmission dynamics of co-endemic Plasmodium vivax and P. falciparum in Ethiopia and prevalence of antimalarial resistant genotypes. PLoS Negl. Trop. Dis. 11, e0005806. https://doi.org/10.1371/journal.pntd.0005806 (2017).
Google Scholar
-
Taylor, A. R. et al. Quantifying connectivity between local Plasmodium falciparum malaria parasite populations using identity by descent. PLoS Genet. 13, e1007065. https://doi.org/10.1371/journal.pgen.1007065 (2017).
Google Scholar
-
Taylor, A. R., Echeverry, D. F., Anderson, T. J. C., Neafsey, D. E. & Buckee, C. O. Identity-by-descent with uncertainty characterises connectivity of Plasmodium falciparum populations on the Colombian-Pacific coast. PLoS Genet. 16, e1009101. https://doi.org/10.1371/journal.pgen.1009101 (2020).
Google Scholar
-
Schewel, K. & Asmamaw, L. B. Migration and development in Ethiopia: Exploring the mechanisms behind an emerging mobility transition. Migr. Stud. 9, 1673–1707. https://doi.org/10.1093/migration/mnab036 (2021).
Google Scholar
-
Price, R. N. et al. Global extent of chloroquine-resistant Plasmodium vivax: A systematic review and meta-analysis. Lancet Infect. Dis. 14, 982–991. https://doi.org/10.1016/S1473-3099(14)70855-2 (2014).
Google Scholar
-
Getachew, S. et al. Chloroquine efficacy for Plasmodium vivax malaria treatment in southern Ethiopia. Malar. J. 14, 1–8 (2015).
Google Scholar
-
Hwang, J. et al. In vivo efficacy of artemether-lumefantrine and chloroquine against Plasmodium vivax: A randomized open label trial in central Ethiopia. PLoS One 8, e63433. https://doi.org/10.1371/journal.pone.0063433 (2013).
Google Scholar
-
Beyene, H. B., Beyene, M. B., Ebstie, Y. A. & Desalegn, Z. Efficacy of chloroquine for the treatment of vivax malaria in northwest Ethiopia. PLoS One 11, e0161483. https://doi.org/10.1371/journal.pone.0161483 (2016).
Google Scholar
-
Assefa, A. et al. Therapeutic efficacies of artemether-lumefantrine and dihydroartemisinin-piperaquine for the treatment of uncomplicated Plasmodium falciparum and chloroquine and dihydroartemisinin-piperaquine for uncomplicated Plasmodium vivax infection in Ethiopia. Malar. J. 21, 359. https://doi.org/10.1186/s12936-022-04350-z (2022).
Google Scholar
-
Gebru-Woldearegai, T., Hailu, A., Grobusch, M. P. & Kun, J. F. Molecular surveillance of mutations in dihydrofolate reductase and dihyropteroate synthase genes of Plasmodium falciparum in Ethiopia. Am. J. Trop. Med. Hyg. 73, 1131–1134 (2005).
Google Scholar
-
ter Kuile, F. O., van Eijk, A. M. & Filler, S. J. Effect of sulfadoxine-pyrimethamine resistance on the efficacy of intermittent preventive therapy for malaria control during pregnancy: A systematic review. JAMA 297, 2603–2616 (2007).
Google Scholar
-
Aponte, J. J. et al. Efficacy and safety of intermittent preventive treatment with sulfadoxine-pyrimethamine for malaria in African infants: A pooled analysis of six randomised, placebo-controlled trials. Lancet 374, 1533–1542. https://doi.org/10.1016/S0140-6736(09)61258-7 (2009).
Google Scholar
-
Dicko, A. et al. Intermittent preventive treatment of malaria provides substantial protection against malaria in children already protected by an insecticide-treated bednet in Mali: A randomised, double-blind, placebo-controlled trial. PLoS Med. 8, e1000407. https://doi.org/10.1371/journal.pmed.1000407 (2011).
Google Scholar
-
Hastings, M. D. & Sibley, C. H. Pyrimethamine and WR99210 exert opposing selection on dihydrofolate reductase from Plasmodium vivax. Proc. Natl. Acad. Sci. 99, 13137–13141 (2002).
Google Scholar
-
Su, X.-Z., Kirkman, L. A., Fujioka, H. & Wellems, T. E. Complex polymorphisms in an ~330 kDa protein are linked to chloroquine-resistant P. falciparum in Southeast Asia and Africa. Cell 91, 593–603 (1997).
Google Scholar
-
Wellems, T. E., Walker-Jonah, A. & Panton, L. J. Genetic mapping of the chloroquine-resistance locus on Plasmodium falciparum chromosome 7. Proc. Natl. Acad. Sci. 88, 3382–3386 (1991).
Google Scholar
-
Verzier, L. H., Coyle, R., Singh, S., Sanderson, T. & Rayner, J. C. Plasmodium knowlesi as a model system for characterising Plasmodium vivax drug resistance candidate genes. PLoS Negl. Trop. Dis. 13, e0007470. https://doi.org/10.1371/journal.pntd.0007470 (2019).
Google Scholar
-
Winzeler, E. A. Longitudinal study of Plasmodium pathogens identifies new loci associated with artemisinin resistance. Genome Biol. 18, 79. https://doi.org/10.1186/s13059-017-1219-x (2017).
Google Scholar
-
Cerqueira, G. C. et al. Longitudinal genomic surveillance of Plasmodium falciparum malaria parasites reveals complex genomic architecture of emerging artemisinin resistance. Genome Biol. 18, 78. https://doi.org/10.1186/s13059-017-1204-4 (2017).
Google Scholar
-
Miller, L. H., Mason, S. J., Clyde, D. F. & McGinniss, M. H. The resistance factor to Plasmodium vivax in blacks: The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 295, 302–304 (1976).
Google Scholar
-
Popovici, J. et al. Amplification of Duffy binding protein-encoding gene allows Plasmodium vivax to evade host anti-DBP humoral immunity. Nat. Commun. 11, 953. https://doi.org/10.1038/s41467-020-14574-9 (2020).
Google Scholar
-
Lo, E. et al. Molecular epidemiology of Plasmodium vivax and Plasmodium falciparum malaria among Duffy-positive and Duffy-negative populations in Ethiopia. Malar. J. 14, 84. https://doi.org/10.1186/s12936-015-0596-4 (2015).
Google Scholar
-
Abate, A. et al. Vivax malaria in Duffy-negative patients shows invariably low asexual parasitaemia: Implication towards malaria control in Ethiopia. Malar. J. 21, 230. https://doi.org/10.1186/s12936-022-04250-2 (2022).
Google Scholar
-
Lo, E. et al. Contrasting epidemiology and genetic variation of Plasmodium vivax infecting Duffy-negative individuals across Africa. Int. J. Infect. Dis. 108, 63–71. https://doi.org/10.1016/j.ijid.2021.05.009 (2021).
Google Scholar
-
Kepple, D. et al. Alternative invasion mechanisms and host immune response to Plasmodium vivax malaria: Trends and future directions. Microorganisms https://doi.org/10.3390/microorganisms9010015 (2020).
Google Scholar
-
Adam, I. et al. An open dataset of Plasmodium vivax genome variation in 1,895 worldwide samples. Wellcome Open Res. 7, 136. https://doi.org/10.12688/wellcomeopenres.17795.1 (2022).
Google Scholar
-
Lacerda, M. V. G. et al. Single-dose tafenoquine to prevent relapse of Plasmodium vivax malaria. N. Engl. J. Med. 380, 215–228. https://doi.org/10.1056/NEJMoa1710775 (2019).
Google Scholar
-
Ethiopia, F. M. O. H. Ethiopia malaria elimination strategic plan: 2021–2025 (2020).
-
Ratcliff, A. et al. Therapeutic response of multidrug-resistant Plasmodium falciparum and P. vivax to chloroquine and sulfadoxine-pyrimethamine in southern Papua, Indonesia. Trans. R. Soc. Trop. Med. Hyg. 101, 351–359. https://doi.org/10.1016/j.trstmh.2006.06.008 (2007).
Google Scholar
-
Phyo, A. P. et al. Dihydroartemisinin-piperaquine versus chloroquine in the treatment of Plasmodium vivax malaria in Thailand: A randomized controlled trial. Clin. Infect. Dis. 53, 977–984. https://doi.org/10.1093/cid/cir631 (2011).
Google Scholar
-
Auburn, S. et al. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 1, 4. https://doi.org/10.12688/wellcomeopenres.9876.1 (2016).
Google Scholar
-
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Google Scholar
-
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. https://doi.org/10.1038/ng.806 (2011).
Google Scholar
-
Manske, M. et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487, 375–379. https://doi.org/10.1038/nature11174 (2012).
Google Scholar
-
Auburn, S. et al. Characterization of within-host Plasmodium falciparum diversity using next-generation sequence data. PLoS One 7, e32891. https://doi.org/10.1371/journal.pone.0032891 (2012).
Google Scholar
-
Zhu, S. J. et al. The origins and relatedness structure of mixed infections vary with local prevalence of P. falciparum malaria. Elife https://doi.org/10.7554/eLife.40845 (2019).
Google Scholar
-
Zhu, S. J., Almagro-Garcia, J. & McVean, G. Deconvolution of multiple infections in Plasmodium falciparum from high throughput sequencing data. Bioinformatics 34, 9–15. https://doi.org/10.1093/bioinformatics/btx530 (2018).
Google Scholar
-
Schaffner, S. F., Taylor, A. R., Wong, W., Wirth, D. F. & Neafsey, D. E. hmmIBD: Software to infer pairwise identity by descent between haploid genotypes. Malar. J. 17, 196. https://doi.org/10.1186/s12936-018-2349-7 (2018).
Google Scholar
-
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. https://doi.org/10.1093/nar/gkw290 (2016).
Google Scholar
-
Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. https://doi.org/10.1093/bioinformatics/btn129 (2008).
Google Scholar
-
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. https://doi.org/10.1101/gr.094052.109 (2009).
Google Scholar
-
Lo, E. et al. Frequent expansion of Plasmodium vivax Duffy Binding Protein in Ethiopia and its epidemiological significance. PLoS Negl. Trop. Dis. 13, e0007222 (2019).
Google Scholar
Acknowledgements
We thank the patients who contributed their samples to the study, the health workers and field teams who assisted with the sample collections, and the staff of the Wellcome Sanger Institute Sample Logistics, Sequencing, and Informatics facilities for their contributions.
Funding
A.M.K. was supported by the Tropical Disease Research Clinical Research and Development Fellowship (award number P20-0004) grant issued by the World Health Organisation Programme for Research and Training in Tropical Diseases (WHO/TDR). The patient sampling and metadata collection for new samples from Ethiopia were supported by The DELTAS Africa Initiative [DELGEME grant 107740/Z/15/Z] awarded to L.G. The study was also supported by the Bill and Melinda Gates Foundation (INV-043618 awarded to S.A.), the National Health and Medical Research Council of Australia (GNT2001083 awarded to S.A.), and the Wellcome Trust (200909 and ICRG GR071614MA Senior Fellowships in Clinical Science to R.N.P.). The whole genome sequencing component of the study was supported by the Medical Research Council and UK Department for International Development (award number M006212) and the Wellcome Trust (award numbers 206194 and 204911).
Author information
Authors and Affiliations
Contributions
A.M.K., R.N.P., L.G. and S.A. conceived and designed the study. A.M.K., E.S., H.T., E.D.B., M.B., R.D.P., S.V.S. and S.A. conducted data analyses. A.M.K., B.E. and A.R. conducted laboratory processing. B.E., A.Assefa, S.G., A.Aseffa, B.P., E.L., R.M., D.Y., F.N., R.N. and L.G. managed patient enrolments and sampling. J.C.R. and D.P.K contributed critical resources for whole genome sequencing. A.M.K. and S.A. wrote the original draft of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Information 1.
Supplementary Information 2.
Supplementary Information 3.
Supplementary Information 4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Kebede, A.M., Sutanto, E., Trimarsanto, H. et al. Genomic analysis of Plasmodium vivax describes patterns of connectivity and putative drivers of adaptation in Ethiopia.
Sci Rep 13, 20788 (2023). https://doi.org/10.1038/s41598-023-47889-w
-
Received: 04 September 2023
-
Accepted: 20 November 2023
-
Published: 27 November 2023
-
DOI: https://doi.org/10.1038/s41598-023-47889-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.