
Congenital disorders
OPG/RANK/RANKL axis relation to cardiac iron-overload in children with transfusion-dependent thalassemia
Aug
Abstract
OPG/RANK/RANKL axis was reportedly involved in initiating various diseases, especially bone and cardiovascular diseases. This study aimed to assess the relationship between some OPG, RANK, and RANKL polymorphisms and alleles and iron-overload-induced cardiomyopathy in children with transfusion-dependent thalassemia (TDT). This study included 80 TDT children and 80 age and sex-matched controls. Real-time PCR was done for rs207318 polymorphism for the OPG gene and rs1805034, rs1245811, and rs75404003 polymorphisms for the RANK gene, and rs9594782 and rs2277438 polymorphisms for the RANKL gene. Cardiac T2* MRI and ejection fraction (EF) were done to assess the myocardial iron status and cardiac function. In this study, there were no significant differences in frequencies of the studied polymorphisms between cases and controls (p > 0.05 in all). In TDT children, OPG rs2073618 (G > C) had a significant relation to myocardial iron overload (p = 0.02). Its C allele had significantly more frequent normal EF than its G allele (p = 0.04). RANK rs75404403 (C > DEL) had a significant relation to cardiac dysfunction (p = 0.02). Moreover, the C allele of that gene had significantly more frequent affected EF than its DEL allele (p = 0.02). The A allele of RANKL rs2277438 (G > A) had significantly less frequent severe cardiac iron overload than the G allele (p = 0.04). In conclusion, the OPG/ RANK/RANKL genes may act as genetic markers for iron-induced cardiomyopathy in TDT children. Some of the studied genes’ polymorphisms and alleles were significantly related to myocardial iron overload and cardiac dysfunction in TDT children.
Introduction
Beta thalassemia syndromes are a group of hereditary disorders characterized by genetic mutations resulting in reduced or absent beta-globin chains. The clinical severity of beta-thalassemia ranges from severe transfusion-dependent anemia in the homozygous state to mild to moderate microcytic anemia in the heterozygous state, depending on the severity of the beta-globin gene mutation and co-inheritance of other genetic determinants1. It has been estimated that one thousand children out of 1.5 million live births are born each year suffering from thalassemia in Egypt. It is reported that the carrier rate in Egypt is between 9 and 10% of the population2
The most critical morbidities in thalassemia are related to iron overload resulting from multiple blood transfusions and enhanced intestinal iron absorption3. After transferrin binding sites are saturated, non–transferrin-bound iron (NTBI) is transported through Ca+2 channels into hepatocytes, cardiac myocytes, and endocrine glands. Reactive oxygen species produced by the metabolism of NTBI contribute to cellular dysfunction and apoptosis1.
When iron starts to accumulate in the liver, the hepatic function remains normal or is slightly affected early in the disease. Patients with thalassemia often develop finely nodular hepatic cirrhosis after several decades4. In addition, iron accumulates in cardiac myocytes, particularly the ventricular walls, which causes left ventricular diastolic dysfunction, subsequent pulmonary hypertension, and eventually right ventricular dilatation and heart failure5,6.
While osteoprotegerin (OPG) is a cytokine of the tumor necrosis factor (TNF) receptor superfamily, receptor activator of nuclear factor kappa-B (RANK)/receptor activator of nuclear factor kappa-B ligand (RANKL) is a receptor-ligand pair of the TNF receptor superfamily. The OPG/RANK/RANKL system is considered the key molecular pathway in bone metabolism7.
The OPG/RANK/RANKL axis was discovered in the late 1990s. Its effect on immunity and dendritic cells and its role in bone homeostasis was verified. Recent studies revealed the contribution of the OPG/RANK/RANKL system to the emergent field of osteoimmunology, organogenesis, and disease conditions including, cancer and rheumatoid arthritis8.
Moreover, studies had linked the OPG/RANK/RANKL axis to many hepatic diseases such as non-alcoholic fatty liver disease9, chronic alcoholic liver disease10, and primary biliary cholangitis10, and other studies had linked it to many cardiovascular diseases12,13,14,15.
We found that the OPG/RANK/RANKL axis is linked to cognitive impairment in children with transfusion-dependent thalassemia (TDT)16.
In this study, we wanted to explore the relationship between genetic variants of OPG, RANK, and RANKL and iron-overload-induced cardiac disease in children with TDT.
Subjects and methods
Study design and participants
This prospective cross-sectional study was conducted at the pediatric department, of Minia University Children and Maternity Hospital, from September 2019 to March 2022.
We analyzed the data of the 80 TDT children who regularly come to follow-up visits in the pediatric hematology clinic at Minia University Children’s Hospital. We also included 80 healthy children as a control group who were age and sex-matched with the TDT children. The study was explained in detail to the parents or legal guardians of the participant children and written informed consents were taken from them. The study was designed respecting the expected ethical aspects. It was performed according to the Declaration of Helsinki 1975, as revised in 2008 and approved by the Institutional Review Board and Medical Ethics Committee of Minia University.
Included children were on regular blood transfusion programs (transfusion-dependent). All patients received repeated blood transfusions (10 ml packed RBCs /kg body weight) every 2–6 weeks to keep their hemoglobin level (Hb) around 9 g/dl after each transfusion. All patients were on deferasirox therapy for at least 12 months before being recruited into the study. Their age ranged between 5 and 16 years, with no sex predilection.
We excluded from our study TDT children who had a history of congenital or acquired heart disease, a history of cardiac surgery, were on cardioprotective drugs or iron chelation therapy other than deferasirox.
Sample calculation
Fisher’s formula for sample size determination was used; n = z2Pq/d2. Where n = desired sample size population; Z = standard normal deviation—set at 1.96 at 95% confidence level. P = proportion of the subjects that present the characteristic. For this study, P will be estimated at 0.5, q = 1 − P. Therefore, the desired sample size was calculated to be n = 42.68. When considering the 20% dropout of study participants, so, the required sample size will be 52.
Baseline clinical assessment
All included children were subjected to detailed medical history taking and thorough clinical examination. We emphasize in children with TDT on the history of the age of their first transfusion, transfusion burden/year (ml/kg/year), history of splenectomy, the average frequency of transfusion, and type and duration of chelation therapy.
Laboratory investigations
Sample collection and biochemical analysis
Six ml of venous blood was withdrawn from each subject by aseptic venipuncture. This sample was divided as follows: Two ml were collected on two vacutainer tubes containing EDTA solutions, one tube was used for CBC assay, by automated cell counter (CelltacES, Nihon Kohden, Germany), and the other for DNA extraction. The other four ml of venous blood were put in a serum separator gel tube, and it was allowed to clot for 30 min at 37 °C before centrifugation for 15 min at 3,500 rpm. The separated serum was used for measurement of serum ferritin and liver function tests, and the remaining serum was stored at −20 °C for further assessment of additional investigations. Liver function tests were assayed using fully automated clinical chemistry auto-analyzer system Konelab 60i (Thermo Electron Incorporation, Finland). Ferritin was assayed by indiko clinical chemistry system, Thermo, Finland.
Molecular analysis
Real-time PCR was done for the following SNPs: rs 9594782 (C > T), 2277438 (G > A) for RANKL polymorphisms, rs 1805034 (C > T), 1245811 (A > G), and 75404003 (C > DEL) for RANK polymorphisms, and rs 207318 (G > C) for OPG polymorphism. It was carried out on DT lite 4 Real-Time PCR System (DNA Technology, Russian) using the following program: one cycle of incubation at 50 °C for 2 min then one cycle for 10 min at 95 °C and lastly 40 cycles of incubation at 95 °C fo 00:15 and then for 1:00 min Results were interpreted on software DT lite 4 7.9
MRI
Liver iron concentration (LIC) and T2* MRI were performed in the Department of Radiology, Minia University Children and Maternity Hospital, using MR Philips ingenia 1.5 Tesla (Philips Medical Systems, Netherlands), ECG & respiratory-gated with dedicated phased array Torso coil using single breath-hold multi-echo gradient echo sequence.
Regarding cardiac MRI (CMRI), the acquired images were post-processed using Region-Based Measurement to calculate: myocardial T2* value: a single short-axis mid-ventricular slice was acquired using a single breath-hold ECG-gated multi-echo dark blood technique. This T2* sequence generated a series of eight images with TEs of 1.5–17.3 ms and spacing of 2.3 ms., then a region of interest (ROI) is put in each image to measure signal intensity. For measurement in the heart, correction for positional changes between breath-holds was made to ensure regions of interest were within the myocardium. In addition,—left ventricular ejection fraction (EF) was measured using standard CMR sequence, and EF < 56% was considered an indicator of cardiac dysfunction17.
Myocardial T2* decay was calculated using manual analysis in an electronic spreadsheet with semi-automated analysis software using Thalassemia tools (a plug-in of CMR tools, Cardiovascular imaging solutions, London, UK).
Results of cardiac T2* were categorized as severe ( 20 ms) myocardial involvement18,19.
Statistical analysis
Data will be coded, entered, and analyzed using SPSS (statistical package for social sciences) version 20. descriptive statistics were calculated and expressed as (mean ± standard deviation (SD), range, median and interquartile range (IQR)). The quantitative variables will be compared using paired t-test or one-way ANOVA. The comparison of qualitative variables will be performed using chi-square test or Fisher’s exact test. p-value < 0.05 was considered statistically significant.
Results
The demographic, clinical and basic laboratory data of the TDT children and controls are represented in Table 1.
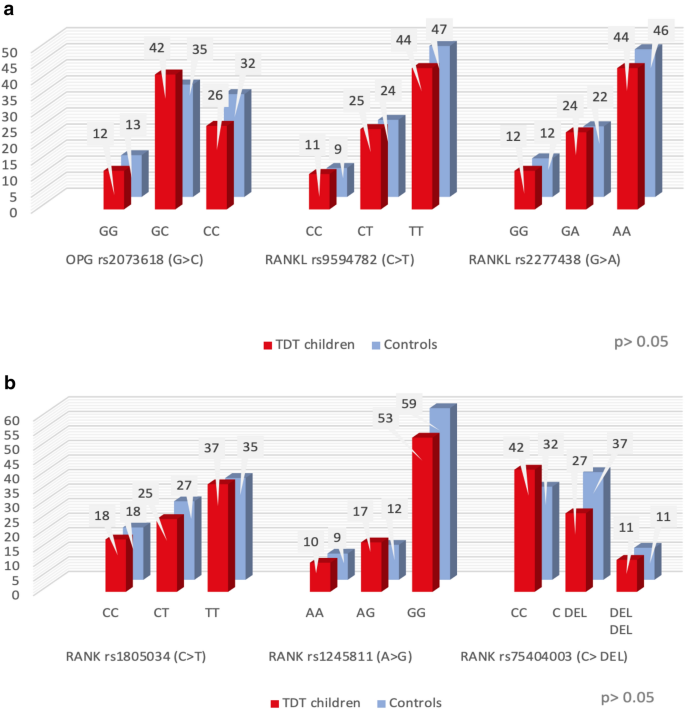
The studied polymorphisms genotypes in patients were not significantly different from those of the controls. (Fig. 1a,b).
(a) and (b) Frequency distribution of the different polymorphisms of the studied genes within the two studied groups.
TDT children had significantly lower T2 and EF than controls, as p < 0.001in all (Table 2).
26 (32.5%) TDT children were categorized as having severe myocardial iron-overload (T2* < 10 ms), while 32 (40%) TDT children were considered as having myocardial dysfunction with an EF < 56%.
OPG rs2073618 (G > C) had a significant relation to myocardial iron overload in the TDT children measured by T2* (p = 0.02, OR = 0.06, 95% CI 0.01–0.67). TDT children with its GG polymorphism were significantly more frequent to have severe iron-overload myocardial involvement than the GC polymorphisms (p = 0.02). Moreover, the A allele of RANKL rs2277438 (G > A) had significantly less frequent T2* < 10 ms than the G allele (p = 0.04, OR = 0.9, 95% CI 0.8–0.94) (Table 3).
RANK rs75404403 (C > DEL) had a significant relation to cardiac dysfunction in TDT children measured by ejection fraction (p = 0.02, OR = 6.1, 95% CI 1.7–22) and children with its CC polymorphism had a significantly more frequent cardiac dysfunction than its C DEL and DEL DEL polymorphisms (p = 0.02 and 0.01 respectively). Moreover, the C allele of that gene had a significantly more frequent impaired ejection fraction than its DEL allele (p = 0.02, OR = 2.8, 95% CI 1.1–6.9). Also, the C allele of the OPG rs2073618 (G > C) gene had a significantly more frequent normal ejection fraction than its G allele (p = 0.04, OR = 2.1, 95% CI 1.0–4.7) (Table 4).
Discussion
Iron overload-induced myocardial dysfunction is TDT’s leading cause of morbidity and mortality20. In this study, TDT children had significantly lower T2* and EF than controls. Previous studies proved that decreased T2* values correlated with myocardial iron overload, deterioration of cardiac functions, and cardiac events in TDT patients21,22,23.
This study aimed to explore the relationship between genetic variants of the OPG/RANK/RANKL axis and iron-overload-induced cardiac abnormalities in children with TDT, as this axis was linked before to oxidative stress-induced disease7, and oxidative stress is the primary mechanism involved in thalassemia iron-overload-induced cardiomyopathy24.
Our study revealed that the OPG rs2073618 (G > C) genotype in TDT children had significant relation with myocardial iron measured by cardiac T2*, and children with its GG polymorphism had significantly more frequent lower T2* than the GC polymorphism. At the same time, the C allele of this gene had a significantly more frequently normal ejection fraction than its G allele.
Our study also found that the A allele of RANKL rs2277438 (G > A) gene had significantly less frequent lower T2* than the G allele. RANK rs75404003 (C > DEL) gene had a significant relation to cardiac function in TDT patients as its CC polymorphism had significantly more frequent cardiac dysfunction than its other two variants. Moreover, the C allele of that gene had a significantly more frequently affected ejection fraction than the DEL allele.
Following our findings, many studies demonstrated the relation of different polymorphisms of this axis with cardiac diseases; Singh et al. study determined that thalassemia patients having RANK rs75404003 (C > DEL), OPG rs2073618 (G > C), and minor C allele of OPG rs2073618, were at high risk for developing left ventricular hypertrophy25. A meta-analysis done by Song et al. in 2016 showed that the OPG rs2073618 genotype is related to cardiovascular disorders such as left ventricle hypertrophy, carotid plaques, and increased risk of stroke26. Also, Straface et al. reported that the CC polymorphism and the C allele of OPG rs2073618 were associated with unstable atherosclerotic plaques27.
Not only was this axis linked to cardiac diseases, but also it was linked to other systemic diseases. RANKL rs2277438 (G > A) had a significant relation to cardiac T2* in our study; other studies had found it to affect bone diseases. Abdi et al. found that its heterozygous GA polymorphism was associated with lower 25(OH) D in postmenopausal Saudi Arabian women28, and Rhee et al. showed its role in vascular calcification and bone metabolism29. The effect of the RANKL rs2277438 (G > A) genotype on both cardiac and bone disease can be explained by an intricate connection between osteogenesis and angiogenesis30. Studies revealed that shear stress and osteoclastic differentiation induce cardiac injury through the expression of OPG/ RANK/RANKL axis genes, in addition to their role in vascular calcification, which is a well-known risk factor for cardiovascular diseases31,32,33.
Iron overload induces free iron radicals, increasing oxidative stress in the myocardium24. The OPG/ RANK/RANKL axis is involved in the endothelial integrity of cardiomyocytes7. and is related to fibroblast growth factors (FGFs), which protect against oxidative stress-related endothelial damage34. Moreover, OPG, RANK, and RANKL genes were proven to have an active role in pathological angiogenesis, inflammation, and cell survival through Vascular endothelial growth factor35. Additionally, studies supported that this axis affects the activity of human fibroblast matrix metalloproteinase (MMP9), which directly relates to myocardial function under pathological conditions36. Confirming the previous studies, Rochette et al. reported that OPG level has a positive association with increased cardiovascular risk and suggested that the increase in OPG levels represents a protective mechanism in response to vascular damage. They concluded that circulating OPG levels can be used as independent prognostic biomarkers of cardiovascular disease in acute or chronic cardiometabolic disorders7. Furthermore, Experimental models of heart failure have confirmed the potential role of OPG in the adaptation of the myocardium to failure as they found a significant increase in mRNA expression of OPG in ischemic and non-ischemic myocardium with heart failure compared with that in subjects without heart failure37.
Conclusion
TDT patients had lower T2* and EF than controls. Furthermore, OPG rs2073618 (G > C), RANK rs75404003 (C > DEL), and the A allele of RANKL rs2277438 (G > A) had significant relation with Myocardial iron overload and cardiac dysfunction occurring in TDT children. Therefore, the OPG/RANK/RANKL pathway impacts iron-overload-induced cardiac dysfunction in TDT children, and the related polymorphisms may act as genetic markers for iron-induced cardiomyopathy in these children.
Study limitations
Assessment of the genetic status of the OPG/RANK/RANKL axis on a broader scale is recommended. In addition, other genes involved in the OPG/RANK/RANKL pathway should also be studied concerning their effect on iron-overload-induced cardiac dysfunction in transfusion-dependent thalassemia patients.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- TDT:
-
Transfusion-dependent thalassemia
- RANK:
-
Receptor activator of nuclear factor-κB
- RANKL:
-
Receptor activator of nuclear factor-κB ligand
- OPG:
-
Osteoprotegerin
- EF:
-
Ejection fraction
- CMRI:
-
Cardiac MRI
References
-
Taher, A. T. & Saliba, A. N. Iron overload in thalassemia: different organs at different rates. Hematol. Am. Soc. Hematol. Educ. Program 2017(1), 265–271. https://doi.org/10.1182/asheducation-2017.1.265 (2017).
Google Scholar
-
El-Shanshory, M. R. et al. Prevalence of iron deficiency anemia and beta thalassemia carriers among relatives of beta thalassemia patients in Nile Delta region, Egypt: A multicenter study. J. Egypt. Public. Health. Assoc. 96, 27 (2021).
Google Scholar
-
Cappellini, M.D., Cohen, A., Porter, J., Taher, A. & Viprakasit, V. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT) 3. Thalassaemia International Federation (2014).
-
Gattermann, N. The treatment of secondary hemochromatosis. Dtsch Arztebl. Int. 106(30), 499. https://doi.org/10.3238/arztebl.2009.0499 (2009).
Google Scholar
-
Kremastinos, D. T. et al. β-thalassemia cardiomyopathy: History, present considerations, and future perspectives. Circ. Heart Fail. 3(3), 451–458 (2010).
Google Scholar
-
Siri-Angkul, N., Xie, L. H., Chattipakorn, S. C. & Chattipakorn, N. Cellular electrophysiology of iron-overloaded cardiomyocytes. Front. Physiol. 9, 1615 (2018).
Google Scholar
-
Rochette, L. et al. The role of osteoprotegerin in the crosstalk between vessels and bone: Its potential utility as a marker of cardiometabolic diseases. Pharmacol. Ther. 1(182), 115–132 (2018).
Google Scholar
-
Walsh, M. C. & Choi, Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol. 5, 511 (2014).
Google Scholar
-
Pacifico, L., Andreoli, G. M., D’Avanzo, M., De Mitri, D. & Pierimarchi, P. Role of osteoprotegerin/receptor activator of nuclear factor kappa B/receptor activator of nuclear factor kappa B ligand axis in nonalcoholic fatty liver disease. World J. Gastroenterol. 24(19), 2073–2082. https://doi.org/10.3748/wjg.v24.i19.2073 (2018).
Google Scholar
-
García-Valdecasas-Campelo, E. et al. Serum osteoprotegerin and RANKL levels in chronic alcoholic liver disease. Alcohol Alcohol. 41(3), 261–266 (2006).
Google Scholar
-
Lleo, A. et al. Quantitation of the Rank–Rankl axis in primary biliary cholangitis. PLoS ONE 11(9), e0159612 (2016).
Google Scholar
-
Venuraju, S. M., Yerramasu, A., Corder, R. & Lahiri, A. Osteoprotegerin as a predictor of coronary artery disease and cardiovascular mortality and morbidity. J. Am. Coll. Cardiol. 55(19), 2049–2061 (2010).
Google Scholar
-
Liu, W. et al. Osteoprotegerin/RANK/RANKL axis in cardiac remodeling due to immuno-inflammatory myocardial disease. Exp. Mol. Pathol. 84(3), 213–217 (2008).
Google Scholar
-
Montagnana, M., Lippi, G., Danese, E. & Guidi, G. C. The role of osteoprotegerin in cardiovascular disease. Ann. Med. 45(3), 254–264 (2013).
Google Scholar
-
Bjerre, M. Osteoprotegerin (OPG) as a biomarker for diabetic cardiovascular complications. Springerplus 2(1), 1–6 (2013).
Google Scholar
-
Mousa, S. O. et al. RANK/RANKL/OPG axis genes relation to cognitive impairment in children with transfusion-dependent thalassemia: A cross-sectional study. BMC Pediat. 22(1), 1–11 (2022).
Google Scholar
-
Meloni, A. et al. Left Ventricular Volumes, Mass and Function normalized to the body surface area, age and gender from CMR in a large cohort of well-treated Thalassemia Major patients without myocardial iron overload. Blood 118(21), 1090 (2011).
Google Scholar
-
Carpenter, J. P. et al. On T2* magnetic resonance and cardiac iron. Circulation 123, 1519–1528 (2011).
Google Scholar
-
Di Tucci, A. A. et al. Myocardial iron overload assessment by T2* magnetic resonance imaging in adult transfusion dependent patients with acquired anemia. Haematologica 93, 1385–1388 (2008).
Google Scholar
-
Gordan, R. et al. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: an update. Heart Fail. Rev. 23(5), 801–816 (2018).
Google Scholar
-
Kirk, P. et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. Circulation 120(20), 1961–1968 (2009).
Google Scholar
-
Majd, Z. et al. Serum ferritin levels correlation with heart and liver MRI and LIC in patients with transfusion-dependent thalassemia. Iran. Red Crescent Med. J. 17, 4 (2015).
Google Scholar
-
Casale, M. et al. Risk factors for endocrine complications in transfusion-dependent thalassemia patients on chelation therapy with deferasirox: A risk assessment study from a multi-center nation-wide cohort. Haematologica 107(2), 467 (2022).
Google Scholar
-
Berdoukas, V., Coates, T. D. & Cabantchik, Z. I. Iron and oxidative stress in cardiomyopathy in thalassemia. Free Radical. Biol. Med. 1(88), 3–9 (2015).
Google Scholar
-
Singh, M. M., Kumar, R., Tewari, S. & Agarwal, S. Investigation of OPG/RANK/RANKL genes as a genetic marker for cardiac abnormalities in Thalassemia major patients. Ann. Hum. Genet. 81(3), 117–124 (2017).
Google Scholar
-
Song, D. H. et al. Relationships of OPG genetic polymorphisms with susceptibility to cardiovascular disease: a meta-analysis. Med. Sci. Monit. Int. Med. J. Experim. Clin. Res. 22, 1223 (2016).
Google Scholar
-
Straface, G. et al. Assessment of the genetic effects of polymorphisms in the osteoprotegerin gene, TNFRSF11B, on serum osteoprotegerin levels and carotid plaque vulnerability. Stroke 42(11), 3022–3028 (2011).
Google Scholar
-
Abdi, S. et al. Association of RANKL and OPG Gene Polymorphism in Arab Women with and without Osteoporosis. Genes 12(2), 200 (2021).
Google Scholar
-
Rhee, E. J. et al. The relationship between receptor activator of nuclear factor-κB ligand (RANKL) gene polymorphism and aortic calcification in Korean women. Endocr. J. 57(6), 541–549 (2010).
Google Scholar
-
Veeriah, V., Paone, R., Chatterjee, S., Teti, A. & Capulli, M. Osteoblasts regulate angiogenesis in response to mechanical unloading. Calcif. Tissue Int. 104(3), 344–354 (2019).
Google Scholar
-
Cho, I. J. et al. Aortic calcification is associated with arterial stiffening, left ventricular hypertrophy, and diastolic dysfunction in elderly male patients with hypertension. J. Hypertens. 33(8), 1633–1641 (2015).
Google Scholar
-
Barbu, C. G. et al. Cardiovascular risk assessment in osteoporotic patients using osteoprotegerin as a reliable predictive biochemical marker. Mol. Med. Rep. 16(5), 6059–6067 (2017).
Google Scholar
-
Liao, C. et al. Shear stress inhibits IL-17A-mediated induction of osteoclastogenesis via osteocyte pathways. Bone 101, 10–20 (2017).
Google Scholar
-
Cao, F. et al. Fibroblast growth factor 21 attenuates calcification of vascular smooth muscle cells in vitro. J. Pharm. Pharmacol. 69(12), 1802–1816 (2017).
Google Scholar
-
Potente, M. & Carmeliet, P. The link between angiogenesis and endothelial metabolism. Annu. Rev. Physiol. 79, 43–66 (2017).
Google Scholar
-
Cao, H., Wang, J., Xi, L., Røe, O.D., Chen, Y. & Wang, D. Dysregulated atrial gene expression of osteoprotegerin/receptor activator of nuclear factor-κB (RANK)/RANK ligand axis in the development and progression of atrial fibrillation. Circ. J. 1110131429 (2011).
-
Ueland, T. et al. Dysregulated osteoprotegerin/RANK ligand/RANK axis in clinical and experimental heart failure. Circulation 111(19), 2461–2468 (2005).
Google Scholar
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
S.Z.S., S.O.M. and M.A.A. participated in the design and planning of the study. M.A.A. has done all the lab work. S.O.M. and A.H.A. were responsible for recruiting the cases. M.A.M. was responsible for performing T2* and EF for the patients. S.O.M. and A.H.A. participated in data collection, analysis of results and preparation of drafts of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Sayed, S.Z., Abd El-Hafez, A.H., Abu El-ela, M.A. et al. OPG/RANK/RANKL axis relation to cardiac iron-overload in children with transfusion-dependent thalassemia.
Sci Rep 13, 12568 (2023). https://doi.org/10.1038/s41598-023-39596-3
-
Received: 09 February 2023
-
Accepted: 27 July 2023
-
Published: 02 August 2023
-
DOI: https://doi.org/10.1038/s41598-023-39596-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.