
Infection
Using next generation antimicrobials to target the mechanisms of infection
Sep
Abstract
The remarkable impact of antibiotics on human health is being eroded at an alarming rate by the emergence of multidrug resistant pathogens. There is a recognised consensus that new strategies to tackle infection are urgently needed to limit the devasting impact of antibiotic resistance on our global healthcare infrastructure. Next generation antimicrobials (NGAs) are compounds that target bacterial virulence factors to disrupt pathogenic potential without impacting bacterial viability. By disabling the key virulence factors required to establish and maintain infection, NGAs make pathogens more vulnerable to clearance by the immune system and can potentially render them more susceptible to traditional antibiotics. In this review, we discuss the developing field of NGAs and how advancements in this area could offer a viable standalone alternative to traditional antibiotics or an effective means to prolong antibiotic efficacy when used in combination.
Introduction
In the early 1900s, infectious diseases were the leading cause of death across almost every age demographic worldwide1,2,3,4. However, during the 20th century there was a dramatic decline in the number of people dying from infectious diseases. This decline can be at least in part, attributed to the advent of antibiotics spearheaded by Sir Alexander Fleming’s discovery of penicillin2,5. The term ‘antibiotic’ was first described in 1941, by Prof Selman Waksman, as a small molecule produced by a microbe that possesses antagonistic properties against the growth of other microbes6. Antibiotics work by inhibiting the growth of bacteria (bacteriostatic) or by killing the bacteria (bactericidal)7. Their mechanisms of action vary but they typically target essential bacterial functions such as transcription, translation, cell wall synthesis and DNA replication. Targeting such essential processes imposes a strong negative selection pressure upon bacteria, driving the evolution of antibiotic resistance8. This has meant that the efficacy of frontline antibiotics is being eroded continually by the spread of transmissible resistance conferring genetic elements and the evolution of multi-drug resistant (MDR) pathogens. This has led to the antibiotic resistance crisis, a major threat to our global healthcare infrastructure and modern medicine. With respect to mortality, the scale of this underreported crisis is akin to other major threats facing humanity such as the climate emergency, with 4.95 million deaths associated with bacterial antimicrobial resistant (AMR) infections in 2019, compared to 5.08 million deaths due to climate change9,10. Worryingly, there is an emerging body of compelling evidence that climate change is exacerbating the AMR crisis, with an increased regional ambient temperature being associated with a higher prevalence of antibiotic resistance11,12.
Our current systems and infrastructure for the clinical development of antibiotics and their transition from the bench to the bedside is failing with an exponential decline in the number of newly developed and approved antibiotics over the last three decades13. The significant costs and time associated with bringing a new class of antibiotic to the market and their lack of financial return has disincentivised the pharmaceutical industry. As a result, most multi-national pharmaceutical companies have shelved their antibiotic development pipelines over the last two decades and many start-ups have folded under these significant pressures. This maelstrom of exits has created a major vulnerability in our healthcare infrastructure driving alarming increases in the number of deaths associated with antibiotic-resistant infections9. The financial burdens associated with treating antibiotic-resistant infections is also a major consideration with the estimated medical cost of one patient with an antibiotic-resistant infection in the US ranging from $18,588 to $29,06914. With the increasing rates of AMR, it is predicted that the annual cost of AMR could rise to $100 trillion by 205015. This is forcing a global rethink of how we bring new antibiotics to market and driving more research into the exploration of alternatives to traditional antibiotics such as phage, vaccines and virulence targeting next-generation antimicrobials (NGAs). Additionally, the repurposing of existing drugs as anti-virulence treatments has gained momentum, providing rapid development with a lower cost, and expanding the range of potential combination therapy options.
NGAs are compounds that have antivirulence properties at concentrations that do not impact bacterial viability, therefore minimising the selective pressure they apply and the probability of resistance evolution. The primary function of virulence factors in an infection context is to allow the pathogen to colonise the host16. Thus, targeting virulence factors disrupts the pathogenic potential of these bacteria making it more difficult for them to colonise the host, making them more vulnerable to clearance by the immune system and potentially rendering them more susceptible to traditional antibiotics (Fig. 1). This review discusses the developing field of NGAs and how advancements in this area could offer a viable standalone alternative to traditional antibiotic use or potentially prolong the efficacy of frontline antibiotics when administered in combination.
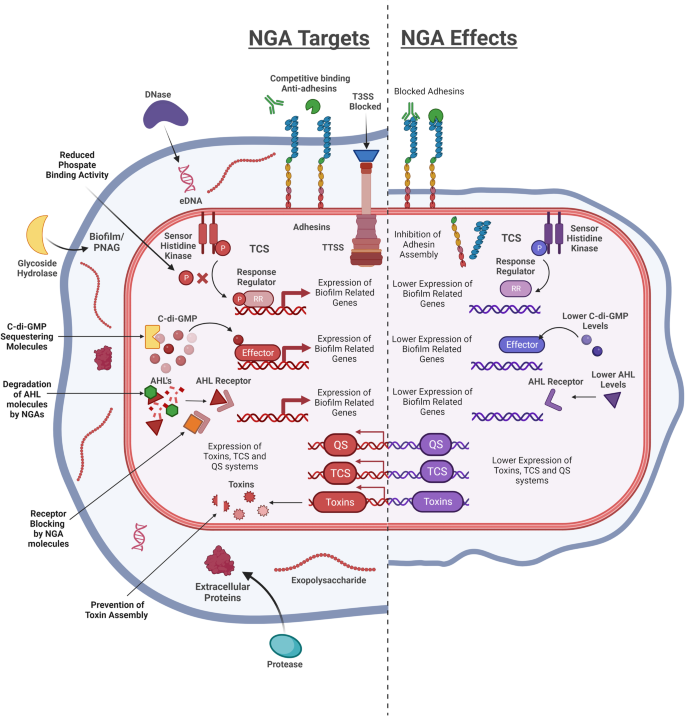
A summary of the different cellular and extracellular targets of NGA (Left) and the impact on different virulent phenotypes (Right).
Colonisation – Disrupting the structural integrity of a biofilm
Biofilms are communities of bacterial cells that adhere to each other or a surface and are encased in a matrix made up of structural components such as polysaccharides, proteins, and extracellular DNA17. Studies have found that ~80% of recurrent or chronic infections are attributed to the formation of bacterial biofilms, highlighting their importance in infection18. The formation of biofilms is a multi-step process that starts with the attachment of bacteria to a biotic or abiotic surface or their aggregation to each other. These micro-colonies then grow and expand with the recruitment of surrounding cells or aggregates and develop into larger three-dimensional community structures with complex nutrient transportation networks. As the biofilm grows, it enters the final stage of its lifecycle, where cells detach from the biofilm and may spread as planktonic cells or aggregates19. Growing in a biofilm provides increased protection from antibiotics, disinfectants and the host immune system. In comparison to cells in a planktonic state, bacteria embedded in biofilms display an increased tolerance to antibiotics by over 10-1000-fold due to poor penetration of antibiotics, heterogeneous transcription and the presence of persister cells20,21,22. These factors are all exacerbated in a polyspecies biofilm where additional behaviours such as cooperation between sensitive and resistant strains or species can occur23,24. Therefore, targeting biofilms is an attractive strategy for the development of NGAs (Table 1). The use of extracellular enzymes that can disrupt biofilms by degrading the structural components of the biofilm matrix is one of the primary strategies for biofilm dispersal. By focusing on the structural integrity of the biofilm, enzymes such as DNase I, PodA and NucB can induce forced dispersal of cells from the biofilm colony and release them into the environment in a more antibiotic susceptible planktonic form25,26,27,28,29,30,31,32,33,34,35.
Targeting extracellular DNA
Extracellular DNA (eDNA) in biofilm functions as structural scaffolding within the matrix and can also modulate aggregation and adhesion to host cells and tissues36. Many studies have shown that the addition of exogenous deoxyribonuclease (DNase) can inhibit biofilm formation in both Gram-negative and Gram-positive bacteria, without impacting bacterial growth37. DNase I cleaves biofilm-associated eDNA resulting in decreased biofilm biomass and an increased antibiotic penetration. This effect has been shown in vitro and in vivo in rat models against a wide range of pathogens including Pseudomonas aeruginosa, Escherichia coli, Acinetobacter baumannii, Staphylococcus aureus and Enterococcus faecalis highlighting the broad-spectrum versatility of this approach26,27,28,29,30,31,38. Indeed, recombinant DNAse I has been used therapeutically for cystic fibrosis (CF) patients for over 20 years as means to decrease the viscoelasticity of sputum slowing the rate of lung function decline. It is likely, based on in vitro data, that the DNAse is also limiting pathogen biofilm formation within the CF lung39. The use of DNases to treat wound biofilms is relatively underexplored in comparison but they have been shown some to disrupt established biofilms and promote healing when administered in combination with silver nanoparticles in vivo40. This disruption of mature biofilms is attributed to the cleavage of eDNA by DNAse, compromising the structural integrity of the biofilm, which in turn allows greater penetration of DNAase enzymes38. The application of DNases to chronic diabetic wounds has also been shown to promote healing, but this is thought to occur through the breakdown of neutrophils extracellular traps (NETs). However, this suggests that a DNase-based chronic wound treatment has the potential to target both host and pathogen factors that are impediments to wound healing41,42.
Rather than targeting the eDNA after it has been integrated into the biofilm matrix, an alternative approach is to inhibit eDNA release. Purified pyocyanin demethylase (PodA) has been shown to inhibit the pyocyanin-dependent release of eDNA into the biofilm matrix, disrupting P. aeruginosa biofilm formation and limiting biofilm aggregate populations32. This approach, however, will not overcome the eDNA that is available through both host and pathogen cell lysis, suggesting that the efficacy of these more targeted approaches may be limited in comparison to exogenous DNAse application43. Another factor to consider is that eDNA has been shown to be protected from DNase degradation by cationic exopolysaccharides, such as the P. aeruginosa polysaccharide Pel, potentially limiting therapeutic efficacy44.
Targeting extracellular proteins
Extracellular proteins are major constituents of the biofilm matrix45. Proteins such as biofilm-associated proteins and DNA-binding proteins play a crucial role in the adhesion, scaffolding and stability of the biofilm matrix46. The integral role of these proteins within the biofilm matrix makes them promising candidates for the development of biofilm dispersal agents. The stable yet highly reactive protease, Proteinase K, has been shown to exhibit biofilm dispersal activity in vitro against several clinically relevant pathogens47,48,49,50,51. Trypsin, a pancreatic serine protease, was found to have a non-cytotoxic biofilm degrading effect on P. aeruginosa52. Similarly, the exogenous application of staphylococcal cysteine proteases Staphopain A (ScpA) and Staphopain B (SspB) have been shown to demonstrate biofilm dispersal abilities against established S. aureus biofilms53,54.
Targeting the immune system towards biofilms associated proteins has been shown to significantly disrupt the structural lattice of eDNA and the overall biofilm. Antisera directed towards DNABII family of proteins such as integration host factor A, IhfA, has been shown to disrupt biofilms formed by each of the high-priority ESKAPE pathogens (Enterococcus faecium, S. aureus, Klebsiella pneumoniae, A. baumannii, P. aeruginosa, Enterobacter spp.,) as well as numerous other clinically relevant pathogens55,56. This approach has also been shown to potentiate DNase induced biofilm damage, antibiotic killing and to increase the capacity of macrophages to kill bacteria55,56. When purified E. coli IHF was used as an immunogen in a chinchilla animal model, with an established biofilm-associated infection, the resultant targeted immune response led to rapid resolution of the infection56. This strategy has also been shown to be effective when targeting polymicrobial biofilms within CF sputum solids57. Humanised monoclonal antibodies directed against DNABII family of proteins have also shown remarkable efficacy to disrupt single and multispecies biofilms and to potentiate antibiotic activity58,59,60.
Targeting extracellular polysaccharides
Secreted extracellular polysaccharides are key components of the biofilm matrix that contribute to the initial establishment and persistence of biofilms61,62. Many studies have demonstrated the efficacy of dispersin B, a glycoside hydrolase produced by Actinobacillus actinomycetemcomitans, against established biofilm of pathogens such as S. aureus, S. epidermidis, A. baumannii, K. pneumoniae, Yersinia pestis and Pseudomonas fluorescens. This glycoside hydrolase degrades the polysaccharide poly(1,6)-N-acetyl-d-glucosamine (PNAG) by hydrolysing the β(1,6) glycosidic linkages30,63. Dispersin B has been used in combination with DNase 1 to limit S. aureus skin colonisation and increase biocide sensitivity in an in vivo porcine model64. Similarly, caspofungin, an antifungal natural product, has been shown to weaken PNAG polymerisation by inhibiting N-acetylglucosamine transferase in S. aureus, resulting in the structure of the biofilm matrix becoming more susceptible to fluoroquinolones in vitro and in vivo in rat models65.
A key consideration with NGAs that are developed to target and disperse biofilms is their potential capacity to send the aggregates and/or planktonic cells into the local microenvironment, potentially facilitating the dissemination of the bacteria to different possible infection sites or triggering bacteraemia25. Therefore, their application must be carefully considered with respect to the type and location of the infection.
Reducing adhesion
The physicochemical properties of the bacterial cell surface and the receptors that decorate it, play a key role in infection, with pili binding to host cell glycoproteins for example often initiating colonisation. Disrupting surface receptor biogenesis has been shown to lead to a decrease in bacterial adhesion to host cells and tissues66. These changes have been shown to occur due to misfolding or an abnormal production of chaperone-usher proteins, that are responsible for the assembly and secretion of fimbrial adhesins. The resultant inhibition of host receptor interactions and alteration in surface charge effectively limits bacterial adhesion67,68,69. This suggests that targeting the assembly of pili, such as Type 1 and P pili found in Escherichia, Salmonella, Yersinia, Pseudomonas, Klebsiella and Haemophilus, may be a promising strategy for preventing bacterial infections via adhesion inhibition70,71.
Small molecules called pilicides have been found to prevent pilus assembly and disrupt formation of the chaperon-usher complex by binding to the active site of the periplasmic chaperones PapD and PapG that are required for the assembly of Type 1 and P pili, and thus preventing bacterial adhesion72,73,74. Sub-inhibitory concentrations of antibiotics like ciprofloxacin and amikacin can also alter the bacterial surface, impairing adhesion to host cells69. Bicyclic 2-pyridones, such as FN075 and BibC6, have demonstrated inhibitory effects on the assembly of curli by preventing polymerisation of the major curli subunit protein CsgA75,76,77,78,79,80. Curli, which are thinner amyloid polymers compared to fimbriae, play a role in adhesion and the formation of biofilms81.
Exploiting carbohydrates that mimic host cell surfaces is a competition-based strategy to prevent bacterial infection, with initially pioneering work by Duguid and Gillis in the 1950s demonstrating the anti-adhesive properties of mannose when applied to E. coli82. This paved the way for the development of a vast array sugar-based inhibitors and glycomimetic compounds that act as anti-adhesives by competitively inhibiting the binding of pathogens to host cells66,82. Multivalent compounds with increased binding avidity and monovalent inhibitors with aglucan moieties have been shown to inhibit uropathogenic E. coli (UPEC) infections by targeting the adhesive subunit FimH83,84. 3′-chloro-4′-(α-d-mannopyranosyloxy) biphenyl-4-carbonitriler, a FimH inhibitor, has shown promising therapeutic potential in the mouse urinary tract infection model, reducing bacterial load in the bladder by almost 1000-fold 3 hours after infection while also displaying favourable pharmacokinetics, such as low toxicity and renal excretion85.
Anti-adhesion antibodies and vaccines are also being explored as strategies to combat bacterial infections. Various approaches have been demonstrated, including immunisation with bacterial adhesins or subunits, immunogenic peptide fragments, or DNA vaccines encoding adhesins66. Targetting the Salmonella enterica serovar Typhi adhesin T2544 using a T2544 antiserum has been shown to enhance the uptake and clearance of bacteria by host macrophages and complement-mediated lysis in mice86. Although antigenic variability could reduce anti-adhesion antibody efficacy, many adhesins are conserved, making them promising vaccine candidates.
Targeting global virulence regulatory pathways
The process of colonisation and pathogenesis is governed by the ability of bacteria to perceive their external environment and the population density. This is regulated by the interconnected systems designated quorum sensing (QS), cyclic di-GMP (CdiGMP) signalling and two component signalling (TCS) systems. As these pathways play diverse roles in controlling bacterial behaviour, disrupting them represents a promising strategy to combat multiple virulence factors at once while typically not impacting bacterial growth directly (Table 2).
Disrupting QS
QS systems are utilised by bacteria as a form of communication to coordinate community phenotypes such as biofilm formation87,88,89. There are three main QS systems. Gram-positive bacteria use specific signalling peptides such as autoinducing peptides (AIPs), and Gram-negative bacteria use N-acylhomoserine lactones (AHLs). Autoinducer-2 (AI-2) is a furanosyl borate diester and is a non-pathogen specific QS molecule. It can facilitate interspecies communication as it is utilised by both Gram-positive and Gram-negative species90. The concentration of autoinducer increases as bacteria grow until a threshold is met. When this point is reached, the cognate response regulators are activated through autoinducer binding and are able to bind to the promoter regions of their target genes, modulating their expression91,92. Given the prevalence of QS systems among pathogens and the key role they play in virulence, targeting QS has become one of the most well-studied strategies for the development of NGAs.
The entire QS regulatory system has been shown to be vulnerable to targeted disruption resulting in virulence attenuation. QS inhibitors can inhibit the expression of components of the QS system or disrupt the interaction between the autoinducer and their cognate receptor proteins. By doing so, these inhibitors can block cell-to-cell communication, biofilm formation and virulence factor production93,94. Salicylic acid and trans-cinnamaldehyde have both been shown to effectively down-regulate the las (LasRI) and rhl (RhlIR) QS systems in P. aeruginosa, in vitro95,96. The specificity of these effects however vary from species to species, with salicylic acid having been shown to stabilise S. aureus biofilms, preventing dispersal97. Several classes of coumarins have also been identified as potent inhibitors of AHL based QS systems, with the simple coumarin molecule being shown to reduce expression of the las, rhl and pqs QS systems in P. aeruginosa and as a result decrease biofilm formation, motility, Type III Secretion System (T3SS) and phenazine production98,99. This activity has been shown to extend to several clinically relevant Gram-positive and Gram-negative bacteria, although the precise mechanism of QS inhibition remains to be uncovered. However, it is worth noting that molecular docking suggests direct interactions with autoinducer synthases100,101. A small-molecule virulence inhibitor, savirin, has been shown to inhibit the Agr QS system in S. aureus by binding to AgrA, preventing its ability to bind to target promoters and ultimately blocking Agr-regulated gene expression, critically at concentrations that do not impact growth102. This molecule has demonstrated efficacy in animal models of biofilm-related S. aureus skin, subcutaneous and prosthetic joint infections by rending the bacteria more susceptible to clearance by skin host defence mechanisms102,103.
Bacteria often compete with other species for the same ecological niche in the natural environment, one strategy that has evolved to increase fitness in this scenario is to disrupt communication between members of the competitor species. The extracellular hydrolysis of autoinducer molecules lowers their local concentration in a process known as quorum quenching (QQ), triggering biofilm dispersal and reduced virulence factor production. QQ enzymes include lactonases, acylases and oxidoreductases and predominantly target AHLs104. Intriguingly, some eukaryotes have been shown to encode QQ enzymes with the capacity to disrupt virulence, in either an example of chance functional promiscuity or perhaps an evolved antivirulence strategy105. Several QQ enzymes have been purified and shown to exhibit potent antivirulence potential against P. aeruginosa in a range of in vivo infection models such as a rat pneumonia model, mouse burn wound model and a mouse pulmonary infection model. The diversity of formulation and delivery of these enzymes also demonstrates their clinical potential with aerosolization, direct application and incorporation into hydrogels and coatings all proving effective delivery mechanisms106,107,108,109.
In the early 21st century, there was considerable excitement about the clinical potential of strategies to target QS, with several pilot clinical trials taking place110,111,112. However, despite the results of these trials being largely positive, the clinical momentum has slowed. This may be impacted due to the emerging evidence that one of the most well studied and targeted QS pathways, the LasRI QS system in P. aeruginosa, is prone to mutations causing loss of function. This indicates that targeting specific QS systems in infection scenarios may not be as effective as originally hoped or as observed in lab adapted strains113,114,115. There has also been some evidence that resistance can evolve to certain classes of QS inhibitor such as furanones116. However, despite these clear limitations, there is still considerable therapeutic promise in targeting QS as a means to tackle the rise in MDR infections.
Blocking CdiGMP signalling
CdiGMP is a secondary messenger molecule produced by diguanylate cyclases (DGCs) and utilised by bacteria to control a broad range of cellular processes, such as biofilm formation, adhesion, motility and virulence117,118. When CdiGMP binds to effector proteins, it has the potential to influence activity, stability, subcellular location, and the proteins’ ability to interact with other proteins117. High levels of CdiGMP are a known trigger of biofilm formation within numerous bacterial species, making approaches to disrupt the regulatory influence of CdiGMP an attractive target for the development of NGAs119. Approaches to disrupt CdiGMP signalling and as a result limit pathogenic potential include the use of synthetic CdiGMP analogs to jam the signalling cascade120,121, disrupting intracellular nucleotide pools122 and the use of DGC active site inhibitors81,123,124. One of the most developed strategies, however, is the use of the nitric oxide to modulate the activity of phosphodiesterases, the enzymes that breakdown intracellular CdiGMP. Exposure to NO has been shown to breakdown and reduce CdiGMP levels by activating CdiGMP-specific phosphodiesterases in bacteria25,125,126,127. Low-dose nitric oxide was also found to cause a significant reduction in P. aeruginosa biofilm aggregates, in CF patients, highlighting the clinical potential of this approach128. As this is an eubacterial secondary messenger, the risks for off target effects needs robust consideration when developing NGAs to target this signalling pathway.
Inhibiting TCS
TCS is utilised by bacteria to sense and respond to changes in the surrounding environment. These systems are critical for bacteria to quickly recognise and adapt to different environmental conditions or threats such as changes in temperature, pH, or nutrient availability129. TCSs are typically composed of two proteins, a sensor kinase, and a cognate response regulator130. The sensor kinase contains a sensor domain that is sensitive to specific environmental signals and undergoes conformational change that activates the kinase domain of the protein. This change then results in the phosphorylation of the histidine residue within the protein. This phosphorylated sensor kinase can then go on to transfer its phosphate group to the response regulator, which contains a DNA-binding domain. Phosphorylation of the response regulator results in a conformational change, which allows for the binding of specific promoter DNA sequences that can then result in the activation or repression of the transcriptional targets131,132. Maprotiline, an FDA-approved tetracyclic antidepressant drug, reduces Francisella novicida biofilm formation through a predicted interaction with the periplasmic sensor domain of histidine kinase, QseC. Treatment of mice infected with F. novicida was shown to improve survival and delay disease onset133. Another QseC inhibitor, the small molecule LED209, was shown to inhibit QseC ligand binding and the resulting autophosphorylation without impacting bacterial viability but critically disabling several virulence mechanisms. It has demonstrated promising efficacy against S. typhimurium and F. tularensis in mouse infection models134,135. Xanthoangelol B, a prenylated chalcone from the plant Angelica keiskei, along with structural derivatives have been shown to directly bind to SaeS, the sensor component of the TCS SaeRS, a major regulator of virulence factor expression in S. aureus136. Mucin glycans have also recently been demonstrated to directly inhibit the TCS GacS-GacA in P. aeruginosa by binding to the antagonistic RetS sensor kinase. This then causes the down regulation of the type 6 secretion system (T6SS) which is associated with bacterial killing137. Despite their role in responding to stimuli, TCS remain a comparatively understudied area for the development of NGAs perhaps due to the essentiality of certain two-component sensors or the potential for host toxicity due to the similarity between kinase domains among bacteria and eukaryotes138,139.
Targeting toxins
Targeting bacterial toxin functionality as a means to limit disease has a long and established history. This approach traces back to the late 19th century when von Behring and Kitasato developed antibody-based antitoxins for Corynebacterium diphtheriae toxin and Clostridium tetani toxin. Their ground-breaking work earned the Nobel Prize for Medicine in 1901140. Over the years, antibody-based antitoxins have made significant progress and have since made their way to clinic. Notably, human monoclonal antibodies targeting Clostridium difficile toxin A and B (actoxumab and bezlotoxumab respectively) having been shown to significantly reduce C. difficile recurrence in several animal models at non-toxic concentrations141,142 and in human clinical trials143,144. However, in phase III clinical trials, only bezlotoxumab alone was shown to reduce C difficile recurrence and as a result was given FDA approval in 2016145. Toxin targeting antibodies have also shown considerable therapeutic promise against other pathogens such as P. aeruginosa, S. aureus and Salmonella spp146,147,148,149,150.
Consequently, secretion systems can be targeted with NGAs at the level of component expression, apparatus assembly, toxin localisation or toxin activity (Table 3). In V. cholera, the transcription of cholera toxin and the toxin coregulated pilus are both regulated by the transcriptional activator ToxT. Through high-throughput screening, the compound 4-[N- (1,8-naphthalimide)]-nbutyric acid (Virstatin) was found to prevent ToxT dimerisation, which is required for promoter binding. In turn, this inhibition blocks the production of the cholera toxin without affecting the growth of the bacteria151,152. The plant phenolic compounds TS027 and TS103 have been shown to impact the regulation of the GacSA-RsmYZ-RsmA-ExsA regulatory pathway in P. aeruginosa which mediates the expression of the toxins of the T3SS153. Salicylidene acylhydrazides have been shown to interfere with the regulation of the T3SS by altering iron availability in bacteria such as Yersinia pseudotuberculosis and Chlamydia trachomatis154. Since this initial discovery, the salicylidene acylhydrazide INP0341 has gone on to show considerable therapeutic promise in corneal, burn and vaginal in vivo models of C. difficile, P. aeruginosa, S. typhimurium, Shigella, C. trachomatis, E. coli infections142,154,155,156,157,158,159,160,161.
Tanshinones, herbal compounds commonly used in traditional Chinese medicine, have been shown to bind directly to components of the P. aeruginosa T3SS needle, preventing needle biogenesis161. Several tanshinones have now been shown to prevent the secretion of T3SS associated toxins to macrophages in vitro and demonstrated efficacy in a murine model of acute pneumonia162. Phenoxyacetamide MBX 1641 was found to bind to the PscF component of the T3SS needle protein in Yersinia pestis and P. aeruginosa, preventing assembly. This inhibitor was found to decrease T3SS mediated cytotoxicity against eukaryotic cells163,164,165. Several small molecule inhibitors of toxin function have been identified and characterised with promising clinical potential. Pseudolipasin A was shown to be an inhibitor of the P. aeruginosa T3SS toxin, ExoU. This inhibitory activity is predicted to occur through the direct binding of this compound to the ExoU catalytic domain166.
An alternative strategy to overcome toxin-mediated virulence is to disrupt the eukaryotic intracellular trafficking of the toxin to its target. Endosome-lysosome acidification is required for the delivery of the C. difficile toxin, TcdB, across the endosomal membrane. This can be effectively inhibited by the general v-ATPase inhibitor bafilomycin A1 as well as several other compounds with lysosomotropic features including the antimalarial drug quinacrine. Preventing the transition of TcdB across the endosomal membrane was sufficient to inhibit TcdB induced cell rounding167,168. The intracellular trafficking of several botulinum neurotoxins has been shown to be inhibited by 4-bromobenzaldehyde N-(2,6-dimethylphenyl) semicarbazone (EGA) effectively reducing neurotoxicity in mouse models168. The cellular toxicity of Shigatoxins STx, STx1, and STx2 is dependent on their retrograde trafficking to their cytosolic target, ribosomes. Several promising compounds have been identified that can disrupt this trafficking and limit toxin activity, including the FDA approved breast cancer chemotherapeutic tamoxifen, which was shown to be a potent inhibitor of STx2 trafficking. Mouse toxicity studies demonstrated that human-approved doses of 10 μM of tamoxifen could significantly improve survival after exposure to a lethal amount of STx1 or STx2169,170,171.
While targeting toxin production may be an effective mechanism to limit acute infection, there is evidence that as a chronic infection develops, toxin production declines, with examples of T3SS inactivating mutations in P. aeruginosa chronic CF and wound isolates172,173,174.
Challenges and future perspectives
The urgent need for novel therapeutic strategies to tackle MDR infections is clear and NGAs represent a promising therapeutical strategy that could overcome key issues like the propensity for resistance evolution associated with traditional antibiotics (Fig. 1). The proposed weaker selection pressure of NGAs, while widely accepted, does not necessarily mean that they are resistance-proof, and the capacity for bacteria to develop mechanisms to overcome their activity is an aspect that needs to be explored in greater detail. NGAs are also expected to typically constitute less interference with mammalian signalling pathways and therefore a reduced toxicity, as they are designed to target virulence pathways that are only found in pathogens, although this obviously is not the case for all NGAs and candidates that target TCSs or host intracellular trafficking in particular need to be robustly screened for off-target effects on the host. There is also the potential that although targeted towards specific pathogens, that NGAs could disrupt the behaviour of commensals within our microbiome, with for example disrupting CdiGMP potentially impacting interspecies competition and the biofilm- forming capacity of commensals within the gut microbiome.
Plant extracts are considered a rich reservoir for bioactive chemicals with high therapeutic potential and have proven to be a rich source of NGA leads. Phytochemicals occupy a chemical space with a far greater structural diversity than synthetic compound libraries and tend to be more ‘drug-like’, with superior ADME/T (absorption, distribution, metabolism, excretion and toxicity) properties. This is due to the evolutionary pressures faced by plants who have endured millennia of intensive selective pressure to develop small molecules that target specific pathways in bacteria to prevent colonisation175. However, a key limitation to the potential of phytochemicals as NGAs is the inherent difficulty in identifying the active molecule within a bioactive plant extract and understanding the specific cellular targets and underlying mechanisms of action, information often necessary for the pre-clinical development of NGAs. This highlights the potential of repurposing previously approved drugs as NGAs, with numerous examples having already been described of drugs having off target antivirulence effects on bacteria133,170,171. Similar potential has been seen with dietary compounds, with artificial sweeteners for example having been recently shown to limit the pathogenicity of several MDR pathogens when used at sub-inhibitory concentrations176. To effectivity stem the tide of MDR pathogens sweeping through our hospitals, it is essential we continue to develop multiple different approaches to tackle these pathogens. Targeting virulence rather than viability is an alternative approach that holds significant therapeutic potential and is likely to have increased clinical importance in the coming years.
References
-
ONS. Causes of Death Over 100 years. (Office for National Statistics, 2021) https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/articles/causesofdeathover100years/2017-09-18.
-
CDC. Achievements in Public Health, 1900-1999: Control of Infectious Diseases. (1999). https://www.cdc.gov/mmwr/preview/mmwrhtml/mm4829a1.htm.
-
Shaw‐Taylor, L. An introduction to the history of infectious diseases, epidemics and the early phases of the long‐run decline in mortality. Econ. Hist. Rev. 73, E1–E19 (2020).
Google Scholar
-
Armstrong, G. L., Conn, L. A. & Pinner, R. W. Trends in infectious disease mortality in the United States during the 20th century’. JAMA 281, 61–66 (1999).
Google Scholar
-
Smith, R. & Coast, J. The economic burden of antimicrobial resistance: Why it is more serious than current studies suggest. (London School of Hygiene & Tropical Medicine, 2013). https://doi.org/10.17037/PUBS.00639028.
-
Clardy, J., Fischbach, M. A. & Currie, C. R. The natural history of antibiotics. Curr. Biol. 19, R437–R441 (2009).
Google Scholar
-
Baquero, F. & Levin, B. R. Proximate and ultimate causes of the bactericidal action of antibiotics. Nat. Rev. Microbiol. 19, 123–132 (2021).
Google Scholar
-
MacLean, R. C. & Millan, San A. The evolution of antibiotic resistance. Science 365, 1082–1083 (2019).
Google Scholar
-
Murray, C. J. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Google Scholar
-
Zhao, Q. et al. Global, regional, and national burden of mortality associated with nonoptimal ambient temperatures from 2000 to 2019: a three-stage modelling study. Lancet Planet Health 5, e415–e425 (2021).
Google Scholar
-
Li, W. et al. Association between antibiotic resistance and increasing ambient temperature in China: an ecological study with nationwide panel data. Lancet Reg. Health West Pac. 30, 100628 (2022).
Google Scholar
-
MacFadden, D. R., McGough, S. F., Fisman, D., Santillana, M. & Brownstein, J. S. Antibiotic resistance increases with local temperature. Nat. Clim. Change. https://www.nature.com/articles/s41558-%20018-0161-6 (2018).
-
Ventola, C. L. The antibiotic resistance crisis. P T 40, 277–283 (2015).
Google Scholar
-
Bartlett, J. G., Gilbert, D. N. & Spellberg, B. Seven ways to preserve the miracle of antibiotics. Clin. Infect. Dis. 56, 1445–1450 (2013).
Google Scholar
-
Dadgostar, P. Antimicrobial resistance: implications and costs. Infect. Drug Resist. 12, 3903–3910 (2019).
Google Scholar
-
Sharma, A. K. et al. Bacterial virulence factors: secreted for survival. Indian J. Microbiol. 57, 1–10 (2017).
Google Scholar
-
Limoli, D. H., Jones, C. J. & Wozniak, D. J. Bacterial extracellular polysaccharides in biofilm formation and function. Microbiol. Spectr. 3, https://doi.org/10.1128/microbiolspec.MB-0011-2014 (2015).
-
Sharma, D., Misba, L. & Khan, A. U. Antibiotics versus biofilm: an emerging battleground in microbial communities. Antimicrob. Resist Infect. Control 8, 76 (2019).
Google Scholar
-
Sauer, K. et al. The biofilm life cycle: expanding the conceptual model of biofilm formation. Nat. Rev. Microbiol. 20, 608–620 (2022).
Google Scholar
-
Uruén, C., Chopo-Escuin, G., Tommassen, J., Mainar-Jaime, R. C. & Arenas, J. Biofilms as promoters of bacterial antibiotic resistance and tolerance. Antibiotics (Basel) 10, 3 (2020).
Google Scholar
-
Pisithkul, T. et al. Metabolic remodeling during biofilm development of Bacillus subtilis. mBio 10, e00623–19 (2019).
Google Scholar
-
Dar, D., Dar, N., Cai, L. & Newman, D. K. Spatial transcriptomics of planktonic and sessile bacterial populations at single-cell resolution. Science 373, eabi4882 (2021).
Google Scholar
-
Frost, I. et al. Cooperation, competition and antibiotic resistance in bacterial colonies. ISME J. 12, 1582–1593 (2018).
Google Scholar
-
Amanatidou, E. et al. Biofilms facilitate cheating and social exploitation of β-lactam resistance in Escherichia coli. npj Biofilms Microbiomes 5, 1–10 (2019).
Google Scholar
-
Rumbaugh, K. P. & Sauer, K. Biofilm dispersion. Nat. Rev. Microbiol. 18, 571–586 (2020).
Google Scholar
-
Sahu, P. K., Iyer, P. S., Oak, A. M., Pardesi, K. R. & Chopade, B. A. Characterization of eDNA from the clinical strain acinetobacter baumannii AIIMS 7 and its role in biofilm formation. Sci. World. J 2012, 973436 (2012).
Google Scholar
-
Rice, K. C. et al. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 104, 8113–8118 (2007).
Google Scholar
-
Thomas, V. C., Thurlow, L. R., Boyle, D. & Hancock, L. E. Regulation of autolysisdependent extracellular DNA release by Enterococcus faecalis extracellular proteases influences biofilm development. J. Bacteriol. 190, 5690–5698 (2008).
Google Scholar
-
Nemoto, K. et al. Effect of Varidase (streptodornase) on biofilm formed by Pseudomonas aeruginosa. Chemotherapy 49, 121–125 (2003).
Google Scholar
-
Waryah, C. B. et al. In vitro antimicrobial efficacy of tobramycin against staphylococcus aureus biofilms in combination with or without DNase I and/or dispersin B: A preliminary investigation. Microb. Drug Resist. 23, 384–390 (2017).
Google Scholar
-
Tetz, V. V. & Tetz, G. V. Effect of extracellular DNA destruction by DNase I on characteristics of forming biofilms. DNA Cell Biol. 29, 399–405 (2010).
Google Scholar
-
Costa, K. C., Glasser, N. R., Conway, S. J. & Newman, D. K. Pyocyanin degradation by a tautomerizing demethylase inhibits Pseudomonas aeruginosa biofilms. Science 355, 170–173 (2017).
Google Scholar
-
Nijland, R., Hall, M. J. & Burgess, J. G. Dispersal of biofilms by secreted, matrix degrading, bacterial DNase. PLoS One 5, e15668 (2010).
Google Scholar
-
Shakir, A., Elbadawey, M. R., Shields, R. C., Jakubovics, N. S. & Burgess, J. G. Removal of biofilms from tracheoesophageal speech valves using a novel marine microbial deoxyribonuclease. Otolaryngol. Head Neck Surg. 147, 509–514 (2012).
Google Scholar
-
Shields, R. C. et al. Efficacy of a marine bacterial nuclease against biofilm forming microorganisms isolated from chronic rhinosinusitis. PLoS One 8, e55339 (2013).
Google Scholar
-
Okshevsky, M. & Meyer, R. L. The role of extracellular DNA in the establishment, maintenance and perpetuation of bacterial biofilms. Crit. Rev. Microbiol 41, 341–352 (2015).
Google Scholar
-
Sharma, K. & Singh, A. P. Antibiofilm effect of DNase against single and mixed species biofilm. Foods 7, 42 (2018).
Google Scholar
-
Tetz, G. V., Artemenko, N. K. & Tetz, V. V. Effect of DNase and antibiotics on biofilm characteristics. Antimicrob. Agents Chemother. 53, 1204–1209 (2009).
Google Scholar
-
Kaplan, J. B. et al. Recombinant human DNase I decreases biofilm and increases antimicrobial susceptibility in staphylococci. J. Antibiot. (Tokyo) 65, 73–77 (2012).
Google Scholar
-
Patel, K. K. et al. Antibiofilm potential of silver sulfadiazine-loaded nanoparticle formulations: a study on the effect of DNase-I on microbial biofilm and wound healing activity. Mol. Pharmaceutics 16, 3916–3925 (2019).
Google Scholar
-
Fadini, G. P. et al. NETosis delays diabetic wound healing in mice and humans. Diabetes 65, 1061–1071 (2016).
Google Scholar
-
Wong, S. L. et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 21, 815–819 (2015).
Google Scholar
-
Whitchurch, C. B. et al. Extracellular DNA required for bacterial biofilm formation. N Y Sci. J. 295, 1487 (2002).
Google Scholar
-
Jennings, L. K. et al. Pseudomonas aeruginosa aggregates in cystic fibrosis sputum produce exopolysaccharides that likely impede current therapies. Cell Rep. 34, 108782 (2021).
Google Scholar
-
Jiao, Y. et al. Characterization of extracellular polymeric substances from acidophilic microbial biofilms. Appl. Environ. Microbiol. 76, 2916–2922 (2010).
Google Scholar
-
Hobley, L., Harkins, C., MacPhee, C. E. & Stanley-Wall, N. R. Giving structure to the biofilm matrix: an overview of individual strategies and emerging common themes. FEMS Microbiol. Rev. 39, 649–669 (2015).
Google Scholar
-
Shukla, S. K. & Rao, T. S. Dispersal of Bap-mediated Staphylococcus aureus biofilm by proteinase K. J. Antibiot. (Tokyo) 66, 55–60 (2013).
Google Scholar
-
Cui, H., Ma, C. & Lin, L. Co-loaded proteinase K/thyme oil liposomes for inactivation of Escherichia coli O157:H7 biofilms on cucumber. Food Funct. 7, 4030–4040 (2016).
Google Scholar
-
Nguyen, U. T. & Burrows, L. L. DNase I and proteinase K impair Listeria monocytogenes biofilm formation and induce dispersal of pre-existing biofilms. Int. J. Food Microbiol. 187, 26–32 (2014).
Google Scholar
-
Fredheim, E. G. A. et al. Biofilm formation by Staphylococcus haemolyticus. J. Clin. Microbiol. 47, 1172–1180 (2009).
Google Scholar
-
Chaignon, P. et al. Susceptibility of staphylococcal biofilms to enzymatic treatments depends on their chemical composition. Appl. Microbiol. Biotechnol. 75, 125–132 (2007).
Google Scholar
-
Banar, M. et al. Evaluation of mannosidase and trypsin enzymes effects on biofilm production of pseudomonas aeruginosa isolated from burn wound infections. PLoS One 11, e0164622 (2016).
Google Scholar
-
Mootz, J. M., Malone, C. L., Shaw, L. N. & Horswill, A. R. Staphopains modulate staphylococcus aureus biofilm integrity. Infect. Immun. 81, 3227–3238 (2013).
Google Scholar
-
Loughran, A. J. et al. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. Microbiologyopen 3, 897–909 (2014).
Google Scholar
-
Novotny, L. A., Amer, A. O., Brockson, M. E., Goodman, S. D. & Bakaletz, L. O. Structural stability of burkholderia cenocepacia biofilms is reliant on eDNA structure and presence of a bacterial nucleic acid binding protein. PLOS ONE 8, e67629 (2013).
Google Scholar
-
Goodman, S. D. et al. Biofilms can be dispersed by focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal. Immunol. 4, 625–637 (2011).
Google Scholar
-
Gustave, J. E., Jurcisek, J. A., McCoy, K. S., Goodman, S. D. & Bakaletz, L. O. Targeting bacterial integration host factor to disrupt biofilms associated with cystic fibrosis. J. Cystic. Fibros. 12, 384–389 (2013).
Google Scholar
-
Novotny, L. A., Jurcisek, J. A., Goodman, S. D. & Bakaletz, L. O. Monoclonal antibodies against DNA-binding tips of DNABII proteins disrupt biofilms in vitro and induce bacterial clearance in vivo. EBioMed. 10, 33–44 (2016).
Google Scholar
-
Jurcisek, J. A., Hofer, L. K., Goodman, S. D. & Bakaletz, L. O. Monoclonal antibodies that target extracellular DNABII proteins or the type IV pilus of nontypeable Haemophilus influenzae (NTHI) worked additively to disrupt 2-genera biofilms. Biofilm 4, 100096 (2022).
Google Scholar
-
Kurbatfinski, N., Goodman, S. D. & Bakaletz, L. O. A humanized monoclonal antibody potentiates killing of diverse biofilm-forming respiratory tract pathogens by antibiotics. Antimicrob. Agents Chemother. 66, e0187721 (2022).
Google Scholar
-
Bales, P. M., Renke, E. M., May, S. L., Shen, Y. & Nelson, D. C. Purification and characterization of biofilm-associated EPS exopolysaccharides from ESKAPE organisms and other pathogens. PLOS ONE 8, e67950 (2013).
Google Scholar
-
Watters, C., Fleming, D., Bishop, D. & Rumbaugh, K. P. Host responses to biofilm. Prog. Mol. Biol. Transl. Sci. 142, 193–239 (2016).
Google Scholar
-
Gawande, P. V. et al. Antibiofilm efficacy of DispersinB® wound spray used in combination with a silver wound dressing. Microbiol. Insights 7, 9–13 (2014).
Google Scholar
-
Kaplan, J. B. et al. Extracellular polymeric substance (EPS)-degrading enzymes reduce staphylococcal surface attachment and biocide resistance on pig skin in vivo. PLOS ONE 13, e0205526 (2018).
Google Scholar
-
Siala, W. et al. The antifungal caspofungin increases fluoroquinolone activity against Staphylococcus aureus biofilms by inhibiting N-acetylglucosamine transferase. Nat. Commun. 7, 13286 (2016).
Google Scholar
-
Krachler, A. M. & Orth, K. Targeting the bacteria–host interface. Virulence 4, 284–294 (2013).
Google Scholar
-
Breines, D. M. & Burnham, J. C. Modulation of Escherichia coli type 1 fimbrial expression and adherence to uroepithelial cells following exposure of logarithmic phase cells to quinolones at subinhibitory concentrations. J. Antimicrob. Chemother. 34, 205–221 (1994).
Google Scholar
-
Dal, S. M. et al. The combination of the SH metabolite of erdosteine (a mucoactive drug) and ciprofloxacin increases the inhibition of bacterial adhesiveness achieved by ciprofloxacin alone. Drugs Exp. Clin. Res. 28, 75–82 (2002).
Google Scholar
-
Wojnicz, D. & Jankowski, S. Effects of subinhibitory concentrations of amikacin and ciprofloxacin on the hydrophobicity and adherence to epithelial cells of uropathogenic Escherichia coli strains. Int. J. Antimicrob. Agents 29, 700–704 (2007).
Google Scholar
-
Nuccio, S.-P. & Bäumler, A. J. Evolution of the chaperone/usher assembly pathway: fimbrial classification goes greek. Microbiol. Mol. Biol. Rev. 71, 551–575 (2007).
Google Scholar
-
Chen, S. L. et al. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. PNAS 103, 5977–5982 (2006).
Google Scholar
-
Svensson, A. et al. Design and evaluation of pilicides: potential novel antibacterial agents directed against uropathogenic Escherichia coli. ChemBioChem 2, 915–918 (2001).
Google Scholar
-
Benz, I. & Schmidt, M. A. AIDA-I, the adhesin involved in diffuse adherence of the diarrhoeagenic Escherichia coli strain 2787 (O126:H27), is synthesized via a precursor molecule. Mol. Microbiol. 6, 1539–1546 (1992).
Google Scholar
-
Berne, C. et al. Adhesins involved in attachment to abiotic surfaces by Gram-negative bacteria, Microbiology spectrum, 3. https://doi.org/10.1128/microbiolspec.MB-0018-2015 (2015).
-
Cegelski, L. et al. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat. Chem. Biol. 5, 913–919 (2009).
Google Scholar
-
Chapman, M. R. et al. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855 (2002).
Google Scholar
-
Connell, H. et al. Bacterial attachment to uro-epithelial cells: mechanisms and consequences. Adv. Dental Res. 11, 50–58 (1997).
Google Scholar
-
Roos, V., Nielsen, E. M. & Klemm, P. Asymptomatic bacteriuria Escherichia coli strains: adhesins, growth and competition. FEMS Microbiol. Letters 262, 22–30 (2006).
Google Scholar
-
Sherlock, O. et al. Novel roles for the AIDA adhesin from diarrheagenic escherichia coli: cell aggregation and biofilm formation. J. Bacteriol. 186, 8058–8065 (2004).
Google Scholar
-
Sherlock, O., Vejborg, R. M. & Klemm, P. The TibA Adhesin/Invasin from enterotoxigenic Escherichia coli is self recognizing and induces bacterial aggregation and biofilm formation. Inf. Immun. 73, 1954–1963 (2005).
Google Scholar
-
Trebino, M. A., Shingare, R. D., MacMillan, J. B. & Yildiz, F. H. Strategies and approaches for discovery of small molecule disruptors of biofilm physiology. Molecules 26, 4582 (2021).
Google Scholar
-
Duguid, J. P. & Gillies, R. R. Fimbrias and adhesive properties in dysentery bacilli. J. Pathol. Bacteriol. 74, 397–411 (1957).
Google Scholar
-
Almant, M. et al. Clustering of Escherichia coli type-1 fimbrial adhesins by using multimeric heptyl α-D-mannoside probes with a carbohydrate core. Chemistry (Weinheim an Der Bergstrasse, Germany) 17, 10029–10038 (2011).
Google Scholar
-
Schierholt, A., Hartmann, M. & Lindhorst, T. K. Bi- and trivalent glycopeptide mannopyranosides as inhibitors of type 1 fimbriae-mediated bacterial adhesion: variation of valency, aglycon and scaffolding. Carbohydr. Res. 346, 1519–1526 (2011).
Google Scholar
-
Kleeb, S. et al. FimH antagonists: bioisosteres to improve the in vitro and in vivo PK/PD profile. J. Med. Chem. 58, 2221–2239 (2015).
Google Scholar
-
Ghosh, S. et al. An adhesion protein of Salmonella enterica serovar Typhi is required for pathogenesis and potential target for vaccine development. Proc. Nat. Acad. Sci. USA 108, 3348–3353 (2011).
Google Scholar
-
Subhadra, B., Kim, D. H., Woo, K., Surendran, S. & Choi, C. H. Control of biofilm formation in healthcare: recent advances exploiting quorum-sensing interference strategies and multidrug efflux pump inhibitors. Materials 11, 1676 (2018).
Google Scholar
-
Bhargava, N., Singh, S. P., Sharma, A., Sharma, P. & Capalash, N. Attenuation of quorum sensing-mediated virulence of Acinetobacter baumannii by Glycyrrhiza glabra flavonoids. Fut. Microbiol. 10, 1953–1968 (2015).
Google Scholar
-
Clemmer, K. M., Bonomo, R. A. & Rather, P. N. Genetic analysis of surface motility in Acinetobacter baumannii. Microbiology 157, 2534 (2011).
Google Scholar
-
Kaur, A., Capalash, N. & Sharma, P. Quorum sensing in thermophiles: prevalence of autoinducer-2 system. BMC Microbiol. 18, 62 (2018).
Google Scholar
-
McCready, A. R., Paczkowski, J. E., Cong, J.-P. & Bassler, B. L. An autoinducerindependent RhlR quorum-sensing receptor enables analysis of RhlR regulation. PLOS Pathogens 15, e1007820 (2019).
Google Scholar
-
Brackman, G. & Coenye, T. Quorum sensing inhibitors as anti-biofilm agents. Curr. Pharm. Des. 21, 5–11 (2015).
Google Scholar
-
Soukarieh, F., Williams, P., Stocks, M. J. & Cámara, M. Pseudomonas aeruginosa Quorum sensing systems as drug discovery targets: current position and future perspectives. J. Med. Chem. (2018).
-
Proctor, C. R., McCarron, P. A. & Ternan, N. G. Furanone quorum-sensing inhibitors with potential as novel therapeutics against Pseudomonas aeruginosa. J. Med. Microbiol. 69, 195–206 (2020).
Google Scholar
-
Ahmed, S. A. K. S. et al. Natural quorum sensing inhibitors effectively downregulate gene expression of Pseudomonas aeruginosa virulence factors. Appl. Microbiol. Biotechnol. 103, 3521–3535 (2019).
Google Scholar
-
Rajkumari, J. et al. Cinnamic acid attenuates quorum sensing associated virulence factors and biofilm formation in Pseudomonas aeruginosa PAO1. Biotechnol. Lett. 40, 1087–1100 (2018).
Google Scholar
-
Dotto, C. et al. Salicylic acid stabilizes Staphylococcus aureus biofilm by impairing the agr quorum-sensing system. Sci. Rep. 11, 2953 (2021).
Google Scholar
-
Zhang, Y. et al. Coumarin reduces virulence and biofilm formation in Pseudomonas aeruginosa by affecting quorum sensing, type III secretion and C-di-GMP levels. Front. Microbiol. 9, 1952 (2018).
Google Scholar
-
Gutiérrez-Barranquero, J. A., Reen, F. J., McCarthy, R. R. & O’Gara, F. Deciphering the role of coumarin as a novel quorum sensing inhibitor suppressing virulence phenotypes in bacterial pathogens. Appl. Microbiol. Biotechnol. 99, 3303–3316 (2015).
Google Scholar
-
Qais, F. A. et al. Coumarin exhibits broad-spectrum antibiofilm and antiquorum sensing activity against gram-negative bacteria: in vitro and in silico investigation. ACS Omega 6, 18823–18835 (2021).
Google Scholar
-
Reen, F. J., Gutiérrez-Barranquero, J. A., Parages, M. L. & O’Gara, F. Coumarin: a novel player in microbial quorum sensing and biofilm formation inhibition. Appl. Microbiol. Biotechnol. 102, 2063–2073 (2018).
Google Scholar
-
Sully, E. K. et al. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 10, e1004174 (2014).
Google Scholar
-
Pant, N. et al. Effect of savirin in the prevention of biofilm-related Staphylococcus aureus prosthetic joint infection. Front. Pharmacol. 13, 989417 (2022).
Google Scholar
-
Rémy, B. et al. Interference in bacterial quorum sensing: a biopharmaceutical perspective. Front. Pharmacol. 9, 203 (2018).
Google Scholar
-
Teiber, J. F. et al. Dominant role of paraoxonases in inactivation of the Pseudomonas aeruginosa quorum-sensing signal N-(3-Oxododecanoyl)-l-Homoserine Lactone. Infect. Immun. 76, 2512 (2008).
Google Scholar
-
Hraiech, S. et al. Inhaled lactonase reduces Pseudomonas aeruginosa quorum sensing and mortality in rat pneumonia. PLOS ONE 9, e107125 (2014).
Google Scholar
-
Sakr, M. M. et al. In vivo evaluation of a recombinant N-acylhomoserine lactonase formulated in a hydrogel using a murine model infected with MDR Pseudomonas aeruginosa clinical isolate, CCASUP2. AMB Express 11, 109 (2021).
Google Scholar
-
Utari, P. D. et al. PvdQ quorum quenching acylase attenuates Pseudomonas aeruginosa virulence in a mouse model of pulmonary infection. Front. Cell Infect. 8, 119 (2018).
Google Scholar
-
Gupta, P., Chhibber, S. & Harjai, K. Efficacy of purified lactonase and ciprofloxacin in preventing systemic spread of Pseudomonas aeruginosa in murine burn wound model. Burns 41, 153–162 (2015).
Google Scholar
-
van Delden, C. et al. Azithromycin to prevent Pseudomonas aeruginosa ventilator associated pneumonia by inhibition of quorum sensing: a randomized controlled trial. J. Intensive Care Med. 38, 1118–1125 (2012).
Google Scholar
-
Smyth, A. R. et al. Garlic as an inhibitor of Pseudomonas aeruginosa quorum sensing in cystic fibrosis—a pilot randomized controlled trial. Pediatr. Pulmonol. 45, 356–362 (2010).
Google Scholar
-
Walz, J. M. et al. Anti-infective external coating of central venous catheters: A randomized, noninferiority trial comparing 5-fluorouracil with chlorhexidine/silver sulfadiazine in preventing catheter colonization*. Crit. Care Med. 38, 2095 (2010).
Google Scholar
-
Smith, E. E. et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 103, 8487–8492 (2006).
Google Scholar
-
Feltner, J. B. et al. LasR variant cystic fibrosis isolates reveal an adaptable quorum-sensing hierarchy in Pseudomonas aeruginosa. mBio 7, e01513–e01516 (2016).
Google Scholar
-
O’Connor, K., Zhao, C. Y., Mei, M. & Diggle, S. P. Frequency of quorum-sensing mutations in Pseudomonas aeruginosa strains isolated from different environments. Microbiology 168, 001265 (2022).
Google Scholar
-
Bové, M., Bao, X., Sass, A., Crabbé, A. & Coenye, T. The quorum-sensing inhibitor furanone c-30 rapidly loses its tobramycin-potentiating activity against Pseudomonas aeruginosa biofilms during experimental evolution. Antimicrob. Agents Chemother. 65, e00413–e00421 (2021).
Google Scholar
-
Jenal, U., Reinders, A. & Lori, C. Cyclic di-GMP: second messenger extraordinaire. Nat. Rev. Microbiol. 15, 271–284 (2017).
Google Scholar
-
Andersen, J. B. et al. Identification of small molecules that interfere with c-di-GMP signaling and induce dispersal of Pseudomonas aeruginosa biofilms. npj Biofilms Microbiomes 7, 1–13 (2021).
Google Scholar
-
Valentini, M. & Filloux, A. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling: Lessons from Pseudomonas aeruginosa and Other Bacteria. J. Biol. Chem. 291, 12547–12555 (2016).
Google Scholar
-
Zhou, J. et al. Potent suppression of c-di-GMP synthesis via I-site allosteric inhibition of diguanylate cyclases with 2’-F-c-di-GMP. Bioorg. Med. Chem. 21, 4396–4404 (2013).
Google Scholar
-
Fernicola, S. et al. Synthesis of triazole-linked analogues of c-di-GMP and their interactions with diguanylate cyclase. J. Med. Chem. 58, 8269–8284 (2015).
Google Scholar
-
Antoniani, D. et al. The immunosuppressive drug azathioprine inhibits biosynthesis of the bacterial signal molecule cyclic-di-GMP by interfering with intracellular nucleotide pool availability. Appl. Microbiol. Biotechnol. 97, 7325–7336 (2013).
Google Scholar
-
Fernicola, S. et al. In silico discovery and in vitro validation of catechol-containing sulfonohydrazide compounds as potent inhibitors of the diguanylate cyclase PleD. J. Bacteriol. 198, 147–156 (2016).
Google Scholar
-
Sambanthamoorthy, K. et al. Identification of small molecules that antagonize diguanylate cyclase enzymes to inhibit biofilm formation. Antimicrob. Agents Chemother. 56, 5202–5211 (2012).
Google Scholar
-
Cutruzzolà, F. & Frankenberg-Dinkel, N. Origin and impact of nitric oxide in pseudomonas aeruginosa biofilms. J. Bacteriol. 198, 55–65 (2015).
Google Scholar
-
Williams, D. E. & Boon, E. M. Towards understanding the molecular basis of nitric oxide-regulated group behaviors in pathogenic bacteria. JIN 11, 205–215 (2019).
Google Scholar
-
Barraud, N., Kelso, M. J., Rice, S. A. & Kjelleberg, S. Nitric oxide: a key mediator of biofilm dispersal with applications in infectious diseases. Curr. Pharm. Des. 21, 31–42 (2015).
Google Scholar
-
Howlin, R. P. et al. Low-dose nitric oxide as targeted anti-biofilm adjunctive therapy to treat chronic Pseudomonas aeruginosa infection in cystic fibrosis. Mol. Ther. 25, 2104–2116 (2017).
Google Scholar
-
Sultan, M., Arya, R. & Kim, K. K. Roles of two-component systems in Pseudomonas aeruginosa virulence. Int. J. Mol. Sci. 22, 12152 (2021).
Google Scholar
-
Brannon, J. R. & Hadjifrangiskou, M. The arsenal of pathogens and antivirulence therapeutic strategies for disarming them. Drug Des. Devel. Ther. 10, 1795–1806 (2016).
Google Scholar
-
Stewart, R. C. Protein histidine kinases: assembly of active sites and their regulation in signaling pathways. Curr. Opin. Microbiol. 13, 133–141 (2010).
Google Scholar
-
Marina, A., Waldburger, C. D. & Hendrickson, W. A. Structure of the entire cytoplasmic portion of a sensor histidine-kinase protein. EMBO J. 24, 4247–4259 (2005).
Google Scholar
-
Dean, S. N. & van Hoek, M. L. Screen of FDA-approved drug library identifies maprotiline, an antibiofilm and antivirulence compound with QseC sensor-kinase dependent activity in Francisella novicida. Virulence 6, 487–503 (2015).
Google Scholar
-
Rasko, D. A. et al. Targeting QseC signaling and virulence for antibiotic development. Science 321, 1078–1080 (2008).
Google Scholar
-
Curtis, M. M. et al. QseC inhibitors as an antivirulence approach for gram-negative pathogens. mBio 5, e02165–14 (2014).
Google Scholar
-
Mizar, P. et al. Total synthesis of xanthoangelol B and its various fragments: toward inhibition of virulence factor production of Staphylococcus aureus. J. Med. Chem. 61, 10473–10487 (2018).
Google Scholar
-
Wang, B. X. et al. Mucin glycans signal through the sensor kinase rets to inhibit virulence-associated traits in Pseudomonas aeruginosa. Curr. Biol. 31, 90–102.e7 (2021).
Google Scholar
-
Tiwari, S. et al. Two-component signal transduction systems of pathogenic bacteria as targets for antimicrobial therapy: an overview’. Front. Microbiol. 8, 1878 (2017).
Google Scholar
-
Chen, H. et al. Recent advances in histidine kinase-targeted antimicrobial agents. Front. Chem. 10, 866392 (2022).
Google Scholar
-
Kantha, S. S. A centennial review; the 1890 tetanus antitoxin paper of von Behring and Kitasato and the related developments. Keio J. Med 40, 35–39 (1991).
Google Scholar
-
Hernandez, L. D. et al. Broad coverage of genetically diverse strains of clostridium difficile by actoxumab and bezlotoxumab predicted by in vitro neutralization and epitope modeling. Antimicrob. Agents Chemother. 59, 1052–1060 (2015).
Google Scholar
-
Yang, Z. et al. Mechanisms of protection against clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect. Immun. 83, 822–831 (2015).
Google Scholar
-
Lowy, I. et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. NEJM 362, 197–205 (2010).
Google Scholar
-
Wilcox, M. H. et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. NEJM 376, 305–317 (2017).
Google Scholar
-
Alonso, C. D. & Mahoney, M. V. Bezlotoxumab for the prevention of Clostridium difficile infection: a review of current evidence and safety profile. Infect Drug Resist 12, 1–9 (2018).
Google Scholar
-
DiGiandomenico, A. et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci. Transl. Med. 6, 262ra155 (2014).
Google Scholar
-
Tkaczyk, C. et al. Targeting alpha toxin and ClfA with a multimechanistic monoclonal-antibody-based approach for prophylaxis of serious Staphylococcus aureus disease. mBio 7, e00528–16 (2016).
Google Scholar
-
Nguyen, T. et al. The structural basis of Salmonella A2B5 toxin neutralization by antibodies targeting the glycan-receptor binding subunits. Cell Rep. 36, 109654 (2021).
Google Scholar
-
Neely, A. N., Holder, I. A., Wiener-Kronish, J. P. & Sawa, T. Passive anti-PcrV treatment protects burned mice against Pseudomonas aeruginosa challenge. Burns 31, 153–158 (2005).
Google Scholar
-
Surewaard, B. G. J. et al. α-toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis. Cell Host Microb. 24, 271–284.e3 (2018).
Google Scholar
-
Hung, D. T., Shakhnovich, E. A., Pierson, E. & Mekalanos, J. J. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310, 670–674 (2005).
Google Scholar
-
Shakhnovich, E. A., Sturtevant, D. & Mekalanos, J. J. Molecular mechanisms of virstatin resistance by non-O1/non-O139 strains of Vibrio cholerae. Mol. Microbiol. 66, 1331–1341 (2007).
Google Scholar
-
Kauppi, A. M., Nordfelth, R., Uvell, H., Wolf-Watz, H. & Elofsson, M. Targeting bacterial virulence: inhibitors of type III secretion in. Yersinia. Chem. Biol. 10, 241–249 (2003).
Google Scholar
-
Uusitalo, P. et al. The salicylidene acylhydrazide INP0341 attenuates Pseudomonas aeruginosa virulence in vitro and in vivo. J. Antibiot. (Tokyo) 70, 937–943 (2017).
Google Scholar
-
Hudson, D. L. et al. Inhibition of type III secretion in Salmonella enterica serovar Typhimurium by small-molecule inhibitors. Antimicrob. Agents Chemother. 51, 2631–2635 (2007).
Google Scholar
-
Veenendaal, A. K. J., Sundin, C. & Blocker, A. J. Small-molecule type III secretion system inhibitors block assembly of the Shigella type III secreton. J. Bacteriol. 191, 563–570 (2009).
Google Scholar
-
Muschiol, S. et al. A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. USA 103, 14566–14571 (2006).
Google Scholar
-
Bailey, L. et al. Small molecule inhibitors of type III secretion in Yersinia block the Chlamydia pneumoniae infection cycle. FEBS Lett. 581, 587–595 (2007).
Google Scholar
-
Tree, J. J. et al. Characterization of the effects of salicylidene acylhydrazide compounds on type III secretion in Escherichia coli O157:H7. Infect. Immun. 77, 4209–4220 (2009).
Google Scholar
-
Anantharajah, A. et al. Salicylidene acylhydrazides and hydroxyquinolines act as inhibitors of type three secretion systems in Pseudomonas aeruginosa by distinct mechanisms. Antimicrob. Agents Chemother. 61, e02566–16 (2017).
Google Scholar
-
Feng, C. et al. Tanshinones: first-in-class inhibitors of the biogenesis of the type 3 secretion system needle of Pseudomonas aeruginosa for antibiotic therapy. ACS Cent. Sci. 5, 1278–1288 (2019).
Google Scholar
-
Aiello, D. et al. Discovery and characterization of inhibitors of Pseudomonas aeruginosa type III secretion. Antimicrob. Agents Chemother. 54, 1988–1999 (2010).
Google Scholar
-
Williams, J. D. et al. Synthesis and structure-activity relationships of novel phenoxyacetamide inhibitors of the Pseudomonas aeruginosa type III secretion system (T3SS). Bioorg. Med. Chem. 23, 1027–1043 (2015).
Google Scholar
-
Bowlin, N. O. et al. Mutations in the Pseudomonas aeruginosa needle protein gene pscF confer resistance to phenoxyacetamide inhibitors of the type III secretion system. Antimicrob. Agents Chemother. 58, 2211–2220 (2014).
Google Scholar
-
Foulkes, D. M. et al. A pipeline to evaluate inhibitors of the Pseudomonas aeruginosa exotoxin U. Biochem. 478, 647–668 (2021).
Google Scholar
-
Tam, J. et al. Small molecule inhibitors of Clostridium difficile toxin b-induced cellular damage. Chem. Biol. 22, 175–185 (2015).
Google Scholar
-
Zhang, J. et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA 111, 13517–13522 (2014).
Google Scholar
-
Azarnia Tehran, D. et al. A novel inhibitor prevents the peripheral neuroparalysis of botulinum neurotoxins. Sci. Rep. 5, 17513 (2015).
Google Scholar
-
Li, D., Selyunin, A. & Mukhopadhyay, S. Targeting the early endosome-to-golgi transport of shiga toxins as a therapeutic strategy. Toxins 12, 342 (2020).
Google Scholar
-
Selyunin, A. S., Hutchens, S., McHardy, S. F. & Mukhopadhyay, S. Tamoxifen blocks retrograde trafficking of Shiga toxin 1 and 2 and protects against lethal toxicosis. Life Sci. Alliance 2, e201900439 (2019).
Google Scholar
-
Savinova, T., Bocharova, Y., Mayanskiy, N. & Chebotar, I. The phenomenon of T3SS inactivation for Pseudomonas aeruginosa strains from a chronic infection locus: do mutations in T3SS-regulators matter? Microbiol. Spectr. 10, e00494–22 (2022).
Google Scholar
-
Jain, M. et al. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. J. Clin. Microbiol. 42, 5229–5237 (2004).
Google Scholar
-
Karash, S., Nordell, R., Ozer, E. A. & Yahr, T. L. Genome sequences of two Pseudomonas aeruginosa isolates with defects in type III secretion system gene expression from a chronic ankle wound infection. Microbiol. Spectr. 9, e0034021 (2021).
Google Scholar
-
McCarthy, R. R. et al. Cyclic-di-GMP regulates lipopolysaccharide modification and contributes to Pseudomonas aeruginosa immune evasion. Nat. Microbiol. 2, 1–10 (2017).
Google Scholar
-
Borges, A. et al. New perspectives on the use of phytochemicals as an emergent strategy to control bacterial infections including biofilms. Molecules 21, 877 (2016).
Google Scholar
-
de Dios, R. et al. Artificial sweeteners inhibit multidrug-resistant pathogen growth and potentiate antibiotic activity. EMBO Mol. Med. 15, e16397 (2023).
Google Scholar
-
Kany, A. M. et al. Tackling Pseudomonas aeruginosa virulence by a hydroxamic acid-based LasB Inhibitor’. ACS Chem. Biol. 13, 2449–2455 (2018).
Google Scholar
-
Bozhkova, S. A. et al. Oligopeptide sortase inhibitor modulates Staphylococcus aureus cell adhesion and biofilm formation. Antibiotics (Basel, Switzerland) 11, 1836 (2022).
Google Scholar
-
Duca, M. et al. Multivalent fucosides targeting β-propeller lectins from lung pathogens with promising anti-adhesive properties’. ACS Chem. Biol. 17, 3515–3526 (2022).
Google Scholar
-
Kumar, L., Chhibber, S., Kumar, R., Kumar, M. & Harjai, K. Zingerone silences quorum sensing and attenuates virulence of Pseudomonas aeruginosa. Fitoterapia 102, 84–95 (2015).
Google Scholar
-
Dong, Y. H., Xu, J. L., Li, X. Z. & Zhang, L. H. AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora. Proc. Natl. Acad. Sci. USA 97, 3526–3531 (2000).
Google Scholar
-
Dong, Y.-H. et al. Quenching quorum-sensing-dependent bacterial infection by an Nacyl homoserine lactonase. Nature 411, 813–817 (2001).
Google Scholar
-
Rémy, B. et al. Lactonase specificity is key to quorum quenching in Pseudomonas aeruginosa. Front. Microbiol. 11, 762 (2020).
Google Scholar
-
Weiland-Bräuer, N. et al. Highly effective inhibition of biofilm formation by the first metagenome-derived AI-2 Quenching enzyme. Front. Microbiol. 7, 1098 (2016).
Google Scholar
-
Nalca, Y. et al. Quorum-sensing antagonistic activities of azithromycin in Pseudomonas aeruginosa PAO1: a global approach. Antimicrob. Agents Chemother. 50, 1680–1688 (2006).
Google Scholar
-
Sedlmayer, F. et al. 5-Fluorouracil blocks quorum-sensing of biofilm-embedded methicillin-resistant Staphylococcus aureus in mice. Nucleic Acids Res. 49, e73 (2021).
Google Scholar
-
Barraud, N. et al. Nitric oxide-mediated dispersal in single- and multi-species biofilms of clinically and industrially relevant microorganisms. Microbial. Biotechnol. 2, 370–378 (2009).
Google Scholar
-
Cai, Y. & Webb, J. S. Optimization of nitric oxide donors for investigating biofilm dispersal response in Pseudomonas aeruginosa clinical isolates. Appl. Microbiol. Biotechnol. 104, 8859–8869 (2020).
Google Scholar
-
Soren, O. et al. Cephalosporin nitric oxide-donor prodrug DEA-C3D disperses biofilms formed by clinical cystic fibrosis isolates of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 75, 117–125 (2020).
Google Scholar
-
Tripathi, A. et al. A defined and flexible pocket explains Aryl substrate promiscuity of the cahuitamycin starter unit-activating enzyme CahJ. Chembiochem 19, 1595–1600 (2018).
Google Scholar
-
Dow, J. M. et al. Biofilm dispersal in Xanthomonas campestris is controlled by cell-cell signaling and is required for full virulence to plants. Proc. Natl. Acad. Sci. USA 100, 10995–11000 (2003).
Google Scholar
-
Davies, D. G. & Marques, C. N. H. A fatty acid messenger is responsible for inducing dispersion in microbial biofilms. J. Bacteriol. 191, 1393–1403 (2009).
Google Scholar
-
Ma, Q., Yang, Z., Pu, M., Peti, W. & Wood, T. K. Engineering a novel c-di-GMP-binding protein for biofilm dispersal. Environ. Microbiol. 13, 631–642 (2011).
Google Scholar
-
Ma, Q., Zhang, G. & Wood, T. K. Escherichia coli BdcA controls biofilm dispersal in Pseudomonas aeruginosa and Rhizobium meliloti. BMC Res. Notes 4, 447 (2011).
Google Scholar
-
Lord, D. M., Baran, A. U., Wood, T. K., Peti, W. & Page, R. BdcA, a protein important for Escherichia coli biofilm dispersal, is a short-chain dehydrogenase/reductase that binds specifically to NADPH. PLoS One 9, e105751 (2014).
Google Scholar
-
Yang, W.-S. et al. A potential substrate binding pocket of BdcA plays a critical role in NADPH recognition and biofilm dispersal. Biochem. Biophys. Res. Commun. 497, 863–868 (2018).
Google Scholar
-
Ching, S. M., Tan, W. J., Chua, K. L. & Lam, Y. Synthesis of cyclic di-nucleotidic acids as potential inhibitors targeting diguanylate cyclase. Bioorg. Med. Chem. 18, 6657–6665 (2010).
Google Scholar
-
Wiggers, H. et al. Identification of anti-inflammatory and anti-hypertensive drugs as inhibitors of bacterial diguanylate cyclases. J. Braz. Chem. Soc. 29, 297–309 (2017).
-
Guarnieri, M. T., Zhang, L., Shen, J. & Zhao, R. The Hsp90 inhibitor radicicol interacts with the ATP-binding pocket of bacterial sensor kinase PhoQ. J. Mol. Biol. 379, 82–93 (2008).
Google Scholar
-
Oi, H. et al. Identification in traditional herbal medications and confirmation by synthesis of factors that inhibit cholera toxin-induced fluid accumulation. Proc. Natl. Acad. Sci. USA 99, 3042–3046 (2002).
Google Scholar
-
Saito, T. et al. Inhibition by apple polyphenols of ADP-ribosyltransferase activity of cholera toxin and toxin-induced fluid accumulation in mice. Microbiol. Immunol. 46, 249–255 (2002).
Google Scholar
-
Hovey, B. T., Verlinde, C. L., Merritt, E. A. & Hol, W. G. Structure-based discovery of a pore-binding ligand: towards assembly inhibitors for cholera and related AB5 toxins. J. Mol. Biol. 285, 1169–1178 (1999).
Google Scholar
-
Hardy, S. J., Holmgren, J., Johansson, S., Sanchez, J. & Hirst, T. R. Coordinated assembly of multisubunit proteins: oligomerization of bacterial enterotoxins in vivo and in vitro. Proc. Natl. Acad. Sci. USA 85, 7109–7113 (1988).
Google Scholar
-
Yang, F. et al. Small-molecule inhibitors suppress the expression of both type III secretion and amylovoran biosynthesis genes in Erwinia amylovora. Mol. Plant Pathol. 15, 44–57 (2014).
Google Scholar
-
Yamazaki, A. et al. Derivatives of plant phenolic compound affect the type III secretion system of Pseudomonas aeruginosa via a GacS-GacA two-component signal transduction system. Antimicrob. Agents Chemother 56, 36–43 (2012).
Google Scholar
-
Zetterström, C. E. et al. The Resveratrol Tetramer (-)-Hopeaphenol Inhibits Type III Secretion in the Gram-Negative Pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa. PLOS ONE 8, e81969 (2013).
Google Scholar
-
Granata, G., Schiavone, F. & Pipitone, G. Bezlotoxumab in patients with a primary Clostridioides difficile infection: a literature review’. Antibiotics 11, 1495 (2022).
Google Scholar
-
Zigangirova, N. A. et al. Fluorothiazinon, a small-molecular inhibitor of T3SS, suppresses Salmonella oral infection in mice. J. Antibiot. 74, 244–254 (2021).
Google Scholar
-
Sheremet, A. B. et al. Small molecule inhibitor of type three secretion system belonging to a Class 2,4-disubstituted-4H-[1,3,4]-thiadiazine-5-ones Improves Survival and Decreases Bacterial Loads in an Airway Pseudomonas aeruginosa Infection in Mice. Biomed. Res. Int. 2018, 5810767 (2018).
Google Scholar
-
Zahid, M. S. H. et al. Suppression of virulence of toxigenic Vibrio cholerae by anethole through the cyclic AMP (cAMP)-cAMP receptor protein signaling system. PLOS ONE 10, e0137529 (2015).
Google Scholar
-
Kelly, C. P. et al. Effect of endogenous clostridioides difficile toxin antibodies on recurrence of C. difficile. Infection’. Clin Infec. Dis. 71, 81–86 (2020).
Google Scholar
-
François, B. et al. Efficacy and safety of suvratoxumab for prevention of Staphylococcus aureus ventilator-associated pneumonia (SAATELLITE): a multicentre, randomised, double-blind, placebo-controlled, parallel-group, phase 2 pilot trial. Lancet Infec. Dis. 21, 1313–1323 (2021).
Google Scholar
-
Tsai, C.-W. & Morris, S. Approval of raxibacumab for the treatment of inhalation anthrax under the US Food and Drug Administration “Animal Rule”. Front. Microbiol. 6 (2015)
-
Xu, W. et al. A systematic review and meta-analysis of preclinical trials testing anti-toxin therapies for B. anthracis infection: A need for more robust study designs and results. PLoS ONE 12, e0182879 (2017).
Google Scholar
-
Greig, S. L. Obiltoxaximab: first global approval. Drugs 76, 823–830 (2016).
Google Scholar
Acknowledgements
We would like to thank Ciaram Staber for critical review of this manuscript. RRMC is supported by a Biotechnology and Biological Sciences Research Council New Investigator Award BB/V007823/1and NERC NE/X010902/1. RRMC is also supported by the Academy of Medical Sciences/the Wellcome Trust/the Government Department of Business, Energy and Industrial Strategy/the British Heart Foundation/Diabetes UK Springboard Award [SBF0061040].
Author information
Authors and Affiliations
Contributions
K.G. and R.R.M.C. conceived and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
Brunel University London has priority patent fillings covering the therapeutic use of artificial sweeteners.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Gadar, K., McCarthy, R.R. Using next generation antimicrobials to target the mechanisms of infection.
npj Antimicrob Resist 1, 11 (2023). https://doi.org/10.1038/s44259-023-00011-6
-
Received: 12 March 2023
-
Accepted: 28 July 2023
-
Published: 22 September 2023
-
DOI: https://doi.org/10.1038/s44259-023-00011-6