
Cardiovascular
Association of Longer Leukocyte Telomere Length With Cardiac Size, Function, and Heart Failure
Jul
Key Points
Question
Is leukocyte telomere length (LTL) associated with alterations in cardiovascular structure and function?
Findings
In this cross-sectional study including 40 459 UK Biobank participants, longer LTL was associated with higher left ventricular mass, larger ventricular and atrial sizes, and higher stroke volumes. Mendelian randomization analysis demonstrated a potential causal genetic association between LTL and left ventricular mass, ventricular size, and left ventricular stroke volume, and longer LTL was associated with a lower risk of incident heart failure after accounting for potential confounders.
Meaning
These findings highlight that modulation of LTL dynamics may have a role in improving cardiovascular structure and function, which could potentially explain the observed lower future risk of heart failure.
Importance
Longer leukocyte telomere length (LTL) is associated with a lower risk of adverse cardiovascular outcomes. The extent to which variation in LTL is associated with intermediary cardiovascular phenotypes is unclear.
Objective
To evaluate the associations between LTL and a diverse set of cardiovascular imaging phenotypes
Design, Setting, and Participants
This is a population-based cross-sectional study of UK Biobank participants recruited from 2006 to 2010. LTL was measured using a quantitative polymerase chain reaction method. Cardiovascular measurements were derived from cardiovascular magnetic resonance using machine learning. The median (IQR) duration of follow-up was 12.0 (11.3-12.7) years. The associations of LTL with imaging measurements and incident heart failure (HF) were evaluated by multivariable regression models. Genetic associations between LTL and significantly associated traits were investigated by mendelian randomization. Data were analyzed from January to May 2023.
Exposure
LTL.
Main Outcomes and Measures
Cardiovascular imaging traits and HF.
Results
Of 40 459 included participants, 19 529 (48.3%) were men, and the mean (SD) age was 55.1 (7.6) years. Longer LTL was independently associated with a pattern of positive cardiac remodeling (higher left ventricular mass, larger global ventricular size and volume, and higher ventricular and atrial stroke volumes) and a lower risk of incident HF (LTL fourth quartile vs first quartile: hazard ratio, 0.86; 95% CI, 0.81-0.91; P = 1.8 × 10−6). Mendelian randomization analysis suggested a potential causal association between LTL and left ventricular mass, global ventricular volume, and left ventricular stroke volume.
Conclusions and Relevance
In this cross-sectional study, longer LTL was associated with a larger heart with better cardiac function in middle age, which could potentially explain the observed lower risk of incident HF.
Introduction
Telomeres are protective caps at the end of chromosomes that progressively shorten with each cell division.1-3 When telomeres reach a critical length, cells enter senescence; hence, telomere length is a marker of cellular replicative capacity and history.2 At the population level, there is a considerable interindividual variation in mean telomere length, usually measured in leukocytes (leukocyte telomere length [LTL]) but also present in other tissues.3 In epidemiological studies, we and others have shown that shorter LTL is associated with risk of incident coronary artery disease (CAD) as well as heart failure (HF).4-6 Mendelian randomization (MR) analyses have strongly suggested that the association of shorter LTL with CAD is genetically causal, although evidence for an association with HF is less certain.4
Cardiac imaging measurements, such as left ventricular mass (LVM), are intermediary phenotypes whose variability has also been shown to influence adverse cardiovascular outcomes, including CAD and HF.7 Two previous studies have investigated the association of LTL with LVM.8,9 The first by Vasan et al8 investigated 850 Framingham Heart study participants, and the second by Kuznetsova and colleagues9 examined 334 volunteers from the Flemish Study on Environment, Genes and Health Outcomes. Both studies used the LVM estimated by measurements from M-mode echocardiography and reported a positive association between LTL and LVM. However, neither study examined whether the association was consistent with a potential causal effect.
UK Biobank (UKB) is a large population cohort established between 2006 and 2010 of participants aged 40 to 69 years at recruitment.10 Participants have been characterized in detail using questionnaires, physical measurements, urinary and plasma biomarker measurements, genomic assays, and longitudinal linkage with multiple health record systems.11 A subset of participants have also undergone cardiovascular magnetic resonance (CMR) scans. We have recently completed a large-scale measurement of LTL in UKB participants and identified a large number of genetic variants associated with LTL, which are useful for potential causal inference (MR analyses).4,12 We have also derived measurements of cardiac structure and function from the CMR scans using automated artificial intelligence–based protocols.13,14 Here, using these data sets, we have examined (1) observational associations between LTL and cardiac morphology, function, and geometry, including LVM, global ventricular volume and size, left ventricular stroke volume (LVSV), right ventricular stroke volume (RVSV), LVM to end-diastolic volume ratio (LVMVR), atrial maximum volume, and atrial emptying volume, (2) the genetic association between LTL and observationally associated CMR measurements using MR, and (3) the potential causal association between LTL and future development of HF.
Participants
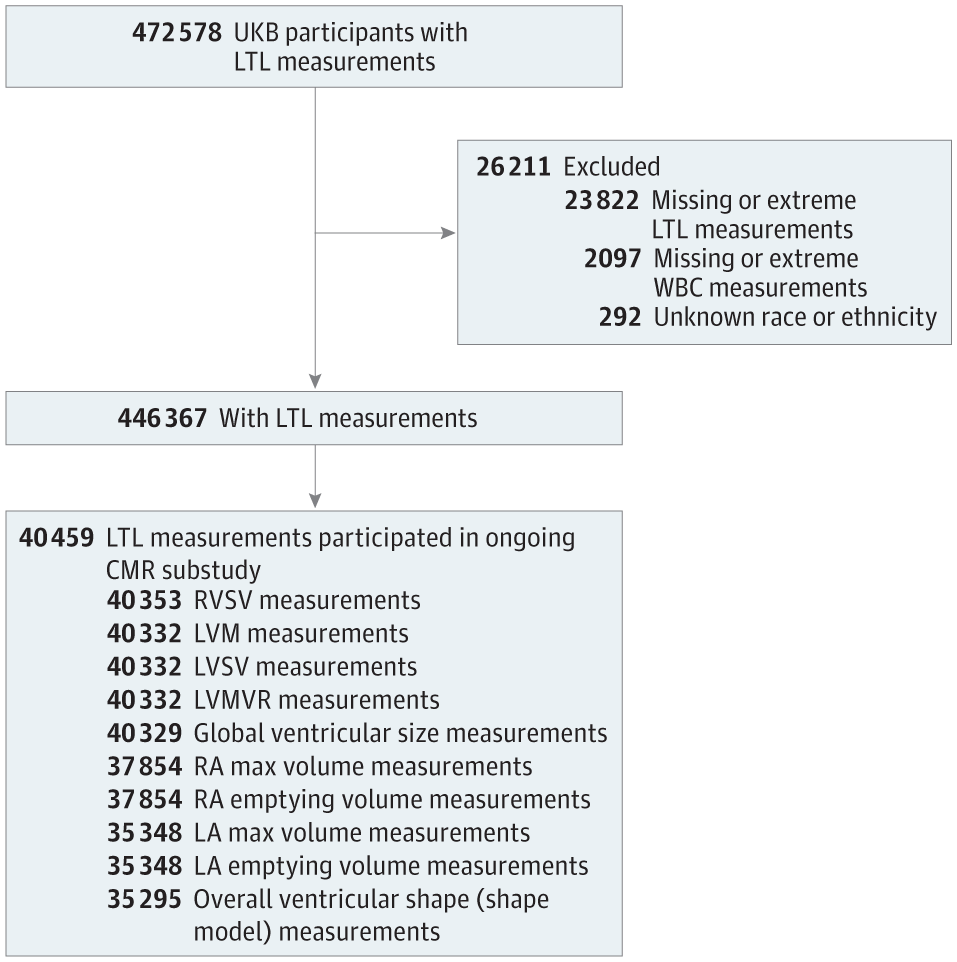
From participants with valid LTL measurements in UKB (n = 474 074),12 we excluded genetically related samples, randomly excluding 1 from each pair based on a kinship coefficient of K > 0.088 and samples with no genetic data or those that failed quality control. We also excluded participants who lacked information on race or ethnicity or white blood cell count, which are both associated with LTL12 and were used together with age and sex to adjust the trait associations. Race and ethnicity data were self-reported by the participants using a touchscreen questionnaire at the assessment center. Available options included Asian or Asian British, Black or Black British, Chinese, White, mixed race, other ethnic group, do not know, and prefer not to answer; those who selected do not know or prefer not to answer were excluded from the study sample. Among the remaining participants with LTL measurements (n = 446 367), 40 459 individuals participated in the ongoing CMR substudy (Figure 1). This study received the overall ethical approval for UKB studies from the NHS National Research Ethics Service on June 17, 2011, which was extended on June 18, 2021. All study participants provided written informed consent. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Measurement of LTL
Details of LTL measurements, including extensive quality checks and the adjustment for technical factors in the UKB participants, have been described previously by our group.12 In brief, LTL was measured as the ratio of telomere repeat copy number (T) relative to that of a single copy gene (S) from the peripheral blood leukocyte DNA, extracted from blood collected at baseline, using a multiplex quantitative polymerase chain reaction method. LTL measurements (T/S ratios) were loge-transformed due to nonnormality (loge[LTL]) and z score standardized for all analyses (UKB field code: 22192).
Derivation of CMR Parameters and Arterial Stiffness
Detailed CMR protocol and analysis methods have been described in prior publications.13-16 Of approximately 500 000 original UKB participants, those living within a reasonable traveling distance to 1 of the 4 imaging assessment centers were invited back for imaging enhancement substudy, with a target sample size of 100 000 individuals. The CMR scans available for the current study (n = 40 000) were obtained a mean (SD) 9.0 (1.7) years after the baseline visit. Segmentation of the left and right ventricular and atrial cavities and left ventricular myocardium were performed by automated machine learning algorithms, as detailed previously.13 Global ventricular volume was defined as the sum of the right and left ventricular end-diastolic volumes. LVSV, RVSV, and atrial stroke volumes were calculated by the difference between respective end-diastolic volume and end-systolic volume. LVMVR was derived by dividing LVM with left ventricular end-diastolic volume. End-diastolic biventricular shape models were obtained, as described previously.17 These models were compiled into a statistical shape atlas through principal component analysis, with principal components capturing the largest sources of variation in cardiac shape among the cohort. Through plotting these principal components, we can estimate biological features that they represent. The first principal component represents the overall size of the heart (higher scores having larger hearts), which was the greatest source of variation in heart shape among individuals (eFigure in Supplement 1).
Statistical Analysis
The descriptive statistics are presented as means with SDs for continuous variables and counts with percentages for categorical variables. The trends across the LTL quartiles were examined by the Cuzick extension of the Wilcoxon rank sum test for continuous variables and the χ2 test for trend for ordinal variables. We removed the confounding influence of chronological age at baseline, white blood cell count, and self-reported race and ethnicity by taking the residuals of loge(LTL) regressed on these variables. Participants with missing data were excluded from the analysis. The associations between loge(LTL) residuals (independent variable) and CMR measures were evaluated in multivariable linear regression models adjusted for age at the imaging visit, sex, height, and weight. Significant associations were additionally adjusted for traditional cardiovascular risk factors (systolic blood pressure, diabetes, dyslipidemia, smoking status, and physical activity expressed in total metabolic equivalent of task minutes per week) to interrogate the potential confounding effects. Given the time lag between LTL sampling and CMR data acquisition, a sensitivity analysis investigating the interaction of the time lag between these 2 dates and LTL was conducted. We sought to identify the association between LTL and incident HF by performing survival analyses using Cox proportional hazards models adjusted for age, sex, body mass index, hypertension, hyperlipidemia, diabetes, and smoking status. We also explored the mediating effect of LTL and LVM on incident HF by introducing an interaction term. The effect sizes were represented by a 1-SD increase in loge(LTL) residuals. Significance was set at P < .05, and all P values were 2-tailed. All analyses were conducted in R version 4.0.2 (The R Foundation).
MR Analysis
To investigate the potential causality and directionality of the associations of LTL with observationally associated imaging traits and with HF, we undertook an MR analysis, using large-scale genome-wide association study data sets.4,14,18 To assess whether the associations between LTL and imaging parameters and HF were consistent with a potential causal association, we used 130 conditionally independent, nonpleiotropic genetic variants that we have recently reported to be associated with LTL in UKB.4
For each analysis, we used the inverse variance–weighted MR method19 allowing for a random effect and also reported the P value for the intercept from MR Egger regression20 as a check for horizontal pleiotropy. As sensitivity analyses, we undertook MR analyses using the weighted median method,21 which is additionally robust in the presence of outliers, and the MR robust adjusted profile score method,22 which overcomes challenges related to measurement error, weak or invalid (due to pleiotropy) measurements, and selection bias (due to weak instrument). We also applied Steiger filtering implemented in the steiger_filtering() function in the R package TwoSampleMR to our genetic instruments, which removed variants that explain more variance in the outcome (ie, imaging measurements or HF) than the exposure (LTL) to minimize the risk of reverse causality. A combination of these approaches provides the best evidence for the presence of a genetic association consistent with a potential causal effect.
Of 40 459 included participants, 19 529 (48.3%) were men, and the mean (SD) age was 55.1 (7.6) years. The baseline characteristics of the study cohort stratified by LTL quartile are presented in Table 1, and characteristics stratified by sex are presented in eTables 1 and 2 in Supplement 1. Individuals in the higher LTL quartiles were more likely to be younger and female with a more favorable traditional cardiovascular risk profile. Most of the study cohort had CMR measurements within normal ranges23; the proportion of LVH was 2% (806 of 40 332). LVM was lower in higher LTL quartiles in the overall cohort, but when stratified by sex, LVM was higher in the higher LTL quartiles. Our study cohort of UKB participants who had CMR assessment were marginally younger, slightly more likely to be male and White, and had a lower prevalence of cardiometabolic risk factors than participants who did not receive CMR examination (eTable 3 in Supplement 1).
Observational Associations Between LTL and Cardiovascular Measurements
After accounting for the differences in age, sex, height, and weight, a positive association was observed between LTL and LVM (β per 1-SD increment in loge[LTL] = 0.47; 95% CI, 0.34-0.60; P = 4.0 × 10−12) (Table 2). Similarly, longer LTL was associated with larger global ventricular volume (β = 1.33; 95% CI, 0.87-1.79; P = 1.8 × 10−8), larger overall ventricular size based on shape modeling (β = 0.01; 95% CI, 0.006-0.02; P = 1.2 × 10−4), higher LVSV (β = 0.35; 95% CI, 0.19-0.50; P = 8.7 × 10−6), higher RVSV (β = 0.34; 95% CI, 0.18-0.50; P = 3.2 × 10−5), larger left atrial maximal volume (β = 0.23; 95% CI, 0.05-0.41; P = .01), and higher left atrial emptying volume (β = 0.12; 95% CI, 0.02-0.23; P = .02). Additional adjustment with cardiovascular risk factors (systolic blood pressure, diabetes, dyslipidemia, smoking status, and physical activity level) slightly attenuated the effect sizes while retaining the statistical significance (Table 2). In contrast, there were no significant associations of LTL with LVMVR, an adverse remodeling phenotype, after adjusting for age, sex, height, and weight. A sensitivity analysis investigating the interaction between LTL and the time lag (between LTL sampling date and imaging visit date) did not find any significant results.
Longitudinal Association Between LTL and Incident HF
Among 406 602 UKB participants with valid LTL measurements free from prevalent cardiovascular diseases, 7827 individuals had incident HF over a median (IQR) follow-up of 12.0 (11.3-12.7) years. In Cox proportional hazards analysis adjusted for age, sex, and other cardiovascular risk factors, longer LTL was associated with a lower future risk of HF (LTL fourth quartile vs first quartile: hazard ratio, 0.86; 95% CI, 0.81-0.91; P = 1.8 × 10−6) (Figure 2). Formal mediation analysis of LTL on the association between LVM (or other imaging traits) and HF was not feasible due to the low event rates in the CMR subcohort (approximately 100 events among approximately 40 000 participants) at this stage. Our exploratory interaction analysis of LTL and LVM on incident HF showed an association with lower risk (interaction HR, 0.87; 95% CI, 0.76-0.99; P = .04).
MR Analyses
Using 130 genetic variants independently associated with LTL as instruments (eTable 4 in Supplement 1), we observed genetic associations of LTL with LVM (β = 0.13; 95% CI, 0.07-0.19; P = .0001), LVSV (β = 0.08; 95% CI, 0.02-0.14; P = .01), global ventricular volume (β = 0.08; 95% CI, 0.02-0.14; P = .01), and biventricular overall size (β = 0.04; 95% CI, 0.0002-0.07; P = .049) from shape model with inverse variance–weighted MR (Figure 3). Other imaging traits and HF did not achieve a statistically significant association with LTL, although the overall effect directions were concordant with observational results. There was no evidence of confounding by directional horizontal pleiotropy. Sensitivity analyses with the weighted median and MR robust adjusted profile score methods gave similar estimates as our primary inverse variance–weighted MR models. Furthermore, Steiger filtering, which removed genetic variants that explain more variance in the outcomes, did not materially alter our findings (eTable 5 in Supplement 1).
Discussion
To our knowledge, this is the first and largest study to investigate the potential causal association between LTL and a comprehensive set of cardiac structure and function, robustly measured with CMR. Our principal findings are that in a middle-aged population (1) longer LTL was associated with higher LVM, larger global ventricular volume and overall size, and higher ventricular and atrial stroke volumes; (2) longer LTL was associated with a lower risk of incident HF even after accounting from traditional cardiovascular risk factors; and (3) the genetic associations between LTL and LVM, LVSV, and global ventricular volume are concordant with the observational results.
Our findings of an association of longer LTL with increased LVM are consistent with 2 previous reports8,9 that assessed LVM using echocardiography and build on these findings. We advanced this insight by highlighting that longer LTL was also associated with larger global ventricular volume and size and higher LVSV. Our finding of better left ventricular systolic function with longer LTL in a general population parallels the data from 2 small prior studies, which reported the associations between shorter LTL and reduced left ventricular ejection fraction in a hypertensive mouse model24 and in a human HF cohort.25 The overall pattern of cardiac morphofunctional differences observed with longer LTL (higher LVM, larger global ventricular volume, static LVMVR, larger atria, and higher ventricular and atrial stroke volumes) closely resembles beneficial balanced myocardial remodeling frequently seen with the physiological adaptation to exercise (eg, athlete heart).26 We also provide compelling genetic evidence, based on multiple MR approaches, that the associations of LTL with LVM, global ventricular volume, and LVSV are consistent with a potential causal association.
The impact of LTL on cardiac structure and function could have clinical relevance. We demonstrated in this work that longer LTL was associated with a reduced observed incidence of HF in UKB (hazard ratio, 0.86; 95% CI, 0.81-0.91; P = 1.8 × 10−6). The MR analysis was nonsignificant (odds ratio per 1-SD longer LTL, 0.96; 95% CI, 0.89-1.03), possibly related to low power. However, no firm conclusion can be drawn based on these data, and future studies using information from larger genome-wide association study are needed. Other studies have shown that LTL is shorter in patients with HF and is associated with poor prognosis.6,27-29 Experimental studies also directly support a role of telomere dynamics in cardiac structure and function. With aging, telomerase knockout mice hearts showed shortening of telomeres, attenuated proliferation and increased apoptosis of cardiomyocytes, and greater cardiac remodeling and left ventricular failure.30,31 On the other hand, enhanced expression of telomerase reverse transcriptase in rat cardiomyocytes preserved telomere length and induced cardiomyocyte proliferation, hypertrophy, and survival.32 While it is recognized that left ventricular hypertrophy and left ventricular dilatation in isolation are associated with adverse outcomes, through access to a more comprehensive set of imaging features, our study demonstrated a more global positive pattern of cardiac remodeling in association with longer LTL, which could explain the lower incidence of HF.
Strengths and Limitations
Our study benefited from several important advantages, including (1) access to the largest sample size to date of LTL data with diverse and accurate cardiovascular imaging measurements using the reference-standard CMR and (2) application of MR for potential causal inference analysis using data from recent large genome-wide association studies. Nevertheless, several limitations need to be acknowledged. First, there is a healthy volunteer selection bias in the UKB, with the participants being older, more affluent, and having a healthier lifestyle with fewer comorbid conditions than the UK general population.33 The imaging substudy cohort is even slightly healthier than the overall UKB cohort. In line with this observation, most of our study cohort had imaging measurements within normal physiological ranges, and the applicability of our findings in disease states leading to left ventricular hypertrophy is uncertain. Second, most of our cohort (97%) is White, which may limit the generalizability of our findings in underrepresented races and ethnicities. Third, telomere length was quantified in blood leukocytes, which may not reflect cell-specific or tissue-specific telomere length. Fourth, the LTL and CMR measurements were obtained at different time points. The impact of this on the findings is uncertain but, if anything, is likely to have blunted the magnitude of the observed associations. Furthermore, our findings from MR, which circumvents the issues of confounding, measurement errors, and reverse causation in observational studies, provide concordant results for the key findings.
Conclusions
In this study, longer LTL was associated with higher LVM, larger global ventricular size, and better cardiac function and a lower risk of incident HF. Further investigations into the prognostic relevance of LTL in adverse cardiac remodeling and the related mechanistic pathways could provide insights into the novel risk stratification approaches and therapeutic targets for HF.
Accepted for Publication: May 31, 2023.
Published Online: July 26, 2023. doi:10.1001/jamacardio.2023.2167
Open Access: This is an open access article distributed under the terms of the CC-BY License. © 2023 Aung N et al. JAMA Cardiology.
Study concept and design: Aung, van Duijvenboden, Raisi-Estabragh, Allara, Wood, Di Angelantonio, Danesh, Young, Harvey, Nelson, Petersen, Samani.
Acquisition, analysis, or interpretation of data: Aung, Wang, van Duijvenboden, Burns, Stoma, Ahmet, Di Angelantonio, Munroe, Codd, Nelson, Petersen, Samani.
Drafting of the manuscript: Aung, van Duijvenboden, Ahmet, Nelson, Samani.
Critical revision of the manuscript for important intellectual content: Aung, Wang, van Duijvenboden, Burns, Stoma, Raisi-Estabragh, Allara, Wood, Di Angelantonio, Danesh, Munroe, Young, Harvey, Codd, Nelson, Petersen.
Statistical analysis: Aung, Wang, van Duijvenboden, Stoma, Wood, Di Angelantonio, Nelson.
Obtained funding: Danesh, Nelson, Petersen, Samani.
Administrative, technical, or material support: Burns, Raisi-Estabragh, Ahmet, Young, Harvey, Codd, Petersen.
Study supervision: Aung, Di Angelantonio, Danesh, Petersen, Samani.
Conflict of Interest Disclosures: Dr Danesh has received grants from the British Heart Foundation during the conduct of the study; serves on scientific advisory boards for AstraZeneca, Novartis, Sanofi, and UK Biobank; is an honorary consultant for Cambridge University Hospital NHS Foundation Trust; and is a member of the Wellcome Trust Sanger Institute, Medical Research Council International Advisory Group, Medical Research Council High Throughput Science ‘Omics Panel, and Wellcome Sanger Institute outside the submitted work. Dr Harvey has received personal fees from UCB, Amgen, and Kyowa Kirin outside the submitted work. Dr Codd has received grants from the Medical Research Council during the conduct of the study. Dr Petersen has received personal fees from Circle Cardiovascular Imaging outside the submitted work. No other disclosures were reported.
Funding/Support: Dr Aung acknowledges the National Institute for Health and Care Research (NIHR) Integrated Academic Training programme which supports his Academic Clinical Lectureship post, and the funding support from the Academy of Medical Sciences Clinical Lecturer Starter Grant. Dr Aung and Dr van Duijvenboden recognize the support from the QMUL Impact Acceleration Account funded by Engineering and Physical Sciences Research Council. Dr Raisi-Estabragh recognizes the NIHR Integrated Academic Training programme, which supports her Academic Clinical Lectureship post, and was also supported by British Heart Foundation Clinical Research Training Fellowship number FS/17/81/33318. Cambridge University investigators are supported by core funding from the British Heart Foundation (RG/13/13/30194; RG/18/13/33946); NIHR Cambridge Biomedical Research Centre (BRC-1215-20014; NIHR203312); and Cambridge BHF Centre of Research Excellence (RE/18/1/34212). Dr Allara was supported by the EU/EFPIA Innovative Medicines Initiative Joint Undertaking BigData@Heart grant 116074 and is currently funded by a British Heart Foundation programme grant (RG/18/13/33946). Dr Wood is part of the BigData@Heart Consortium, funded by the Innovative Medicines Initiative-2 Joint Undertaking under grant agreement No 116074. Dr Wood is supported by the British Heart Foundation Turing Cardiovascular Data Science Award (BCDSA100005). Dr Danesh holds a British Heart Foundation Professorship and a NIHR Senior Investigator Award. Dr Harvey acknowledges support from the UK Medical Research Council (MC_PC_21003; MC_PC_21001) and NIHR Southampton Biomedical Research Centre, University of Southampton, and University Hospital Southampton. Drs Petersen and Munroe acknowledge support from the NIHR Biomedical Research Centre at Barts. Dr Petersen acknowledges the British Heart Foundation for funding the manual analysis to create a cardiovascular magnetic resonance imaging reference standard for the UK Biobank imaging resource in 5000 CMR scans. Dr Petersen acknowledges support from the SmartHeart EPSRC programme grant. Telomere length measurements were funded by the UK Medical Research Council, Biotechnology and Biological Sciences Research Council, and British Heart Foundation through MRC grant MR/M012816/1 to Dr Samani. Dr Nelson is funded by the British Heart Foundation (SP/16/4/32697). Drs Wang, Codd, Nelson, and Samani are supported by the National Institute for Health and Care Research Leicester Cardiovascular Biomedical Research Centre (BRC-1215-20010). Cambridge University investigators are supported by the British Heart Foundation (RG/13/13/30194; RG/18/13/33946), Health Data Research UK, NIHR Cambridge BRC (BRC-1215-20014), NIHR Blood and Transplant Research Unit in Donor Health, and Genomics (NIHR BTRU-2014-10024) and MRC (MR/L003120/1). Dr Danesh holds a BHF Personal Professorship and NIHR Senior Investigator Award.
Role of the Funder/Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Disclaimer: The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Data Sharing Statement: See Supplement 2.
Additional Contributions: This research has been conducted using the UK Biobank Resource under Application 2964. We thank all UK Biobank participants and staff.
References
PC, Heydon
EE, Kaptoge
S, Butterworth
AS, Thompson
A, Willeit
P. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. BMJ. 2014;349:g4227. doi:10.1136/bmj.g4227PubMedGoogle ScholarCrossref
P, van der Steege
G, de Boer
RA,
et al; MERIT-HF Study Group. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J Am Coll Cardiol. 2007;49(13):1459-1464. doi:10.1016/j.jacc.2007.01.027PubMedGoogle ScholarCrossref
G, Gottdiener
JS, Chinali
M, Maurer
MS. Left ventricular mass predicts heart failure not related to previous myocardial infarction: the Cardiovascular Health Study. Eur Heart J. 2008;29(6):741-747. doi:10.1093/eurheartj/ehm605PubMedGoogle ScholarCrossref
T, Codd
V, Brouilette
S,
et al. Association between left ventricular mass and telomere length in a population study. Am J Epidemiol. 2010;172(4):440-450. doi:10.1093/aje/kwq142PubMedGoogle ScholarCrossref
V, Denniff
M, Swinfield
C,
et al. A major population resource of 474,074 participants in UK Biobank to investigate determinants and biomedical consequences of leukocyte telomere length. medRxiv. Preprint posted online March 24, 2021. doi:10.1101/2021.03.18.21253457Google Scholar
N, Vargas
JD, Yang
C,
et al. Genome-wide analysis of left ventricular image-derived phenotypes identifies fourteen loci associated with cardiac morphogenesis and heart failure development. Circulation. 2019;140(16):1318-1330. doi:10.1161/CIRCULATIONAHA.119.041161PubMedGoogle ScholarCrossref
L, Hann
E, Lukaschuk
E,
et al. Automated localization and quality control of the aorta in cine CMR can significantly accelerate processing of the UK Biobank population data. PLoS One. 2019;14(2):e0212272. doi:10.1371/journal.pone.0212272PubMedGoogle ScholarCrossref
C, Gilbert
K, Lee
AM,
et al. Right ventricular shape and function: cardiovascular magnetic resonance reference morphology and biventricular risk factor morphometrics in UK Biobank. J Cardiovasc Magn Reson. 2019;21(1):41. doi:10.1186/s12968-019-0551-6PubMedGoogle ScholarCrossref
S, Henry
A, Roselli
C,
et al; Regeneron Genetics Center. Genome-wide association and mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11(1):163. doi:10.1038/s41467-019-13690-5PubMedGoogle ScholarCrossref
J, Davey Smith
G, Burgess
S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512-525. doi:10.1093/ije/dyv080PubMedGoogle ScholarCrossref
J, Davey Smith
G, Haycock
PC, Burgess
S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304-314. doi:10.1002/gepi.21965PubMedGoogle ScholarCrossref
Q, Wang
J, Hemani
G, Bowden
J, Small
DS. Statistical inference in two-sample summary-data mendelian randomization using robust adjusted profile score. arXiv. Preprint posted online January 1, 2019. doi:10.48550/arXiv.1801.09652Google Scholar
SE, Aung
N, Sanghvi
MM,
et al. Reference ranges for cardiac structure and function using cardiovascular magnetic resonance (CMR) in Caucasians from the UK Biobank population cohort. J Cardiovasc Magn Reson. 2017;19(1):18. doi:10.1186/s12968-017-0327-9PubMedGoogle ScholarCrossref
M, Dörschmann
H, Khraisat
S,
et al. Telomere shortening in hypertensive heart disease depends on oxidative DNA damage and predicts impaired recovery of cardiac function in heart failure. Hypertension. 2022;79(10):2173-2184. doi:10.1161/HYPERTENSIONAHA.121.18935PubMedGoogle ScholarCrossref
VG, Mateo Leach
I, Kjekshus
J,
et al. Telomere length and outcomes in ischaemic heart failure: data from the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Eur J Heart Fail. 2015;17(3):313-319. doi:10.1002/ejhf.237PubMedGoogle ScholarCrossref
A, Franco
S, Zacheo
A,
et al. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003;22(1):131-139. doi:10.1093/emboj/cdg013PubMedGoogle ScholarCrossref
LSM, Oeseburg
H, de Boer
RA, van Gilst
WH, van Veldhuisen
DJ, van der Harst
P. Telomere biology in cardiovascular disease: the TERC−/− mouse as a model for heart failure and ageing. Cardiovasc Res. 2009;81(2):244-252. doi:10.1093/cvr/cvn337PubMedGoogle ScholarCrossref
C, Bernardes de Jesus
B, Serrano
R,
et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat Commun. 2014;5:5863. doi:10.1038/ncomms6863PubMedGoogle ScholarCrossref
A, Littlejohns
TJ, Sudlow
C,
et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol. 2017;186(9):1026-1034. doi:10.1093/aje/kwx246PubMedGoogle ScholarCrossref